

EGFR Regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1
- PMID: 34725466
- PMCID: PMC8560880
- DOI: 10.1038/s42003-021-02744-4
EGFR Regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1
Abstract
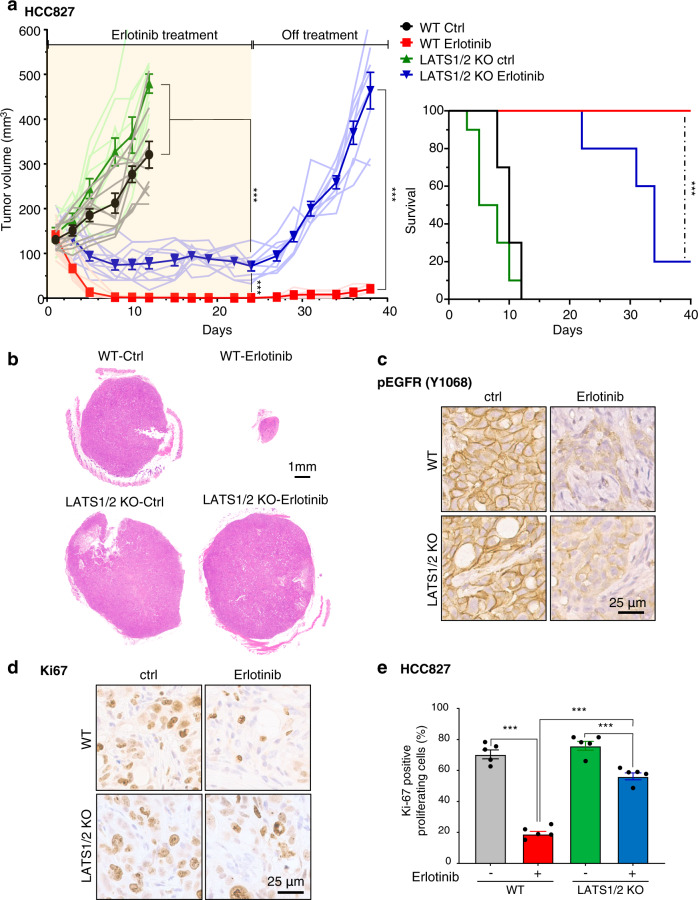
The Hippo pathway is frequently dysregulated in cancer, leading to the unrestrained activity of its downstream targets, YAP/TAZ, and aberrant tumor growth. However, the precise mechanisms leading to YAP/TAZ activation in most cancers is still poorly understood. Analysis of large tissue collections revealed YAP activation in most head and neck squamous cell carcinoma (HNSCC), but only 29.8% of HNSCC cases present genetic alterations in the FAT1 tumor suppressor gene that may underlie persistent YAP signaling. EGFR is overexpressed in HNSCC and many other cancers, but whether EGFR controls YAP activation is still poorly understood. Here, we discover that EGFR activates YAP/TAZ in HNSCC cells, but independently of its typical signaling targets, including PI3K. Mechanistically, we find that EGFR promotes the phosphorylation of MOB1, a core Hippo pathway component, and the inactivation of LATS1/2 independently of MST1/2. Transcriptomic analysis reveals that erlotinib, a clinical EGFR inhibitor, inactivates YAP/TAZ. Remarkably, loss of LATS1/2, resulting in aberrant YAP/TAZ activity, confers erlotinib resistance on HNSCC and lung cancer cells. Our findings suggest that EGFR-YAP/TAZ signaling plays a growth-promoting role in cancers harboring EGFR alterations, and that inhibition of YAP/TAZ in combination with EGFR might be beneficial to prevent treatment resistance and cancer recurrence.
© 2021. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: J.S.G. is a member of the advisory board of Oncoceutics, Domain Therapeutics, and Vividion. The remaining authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
