

Single-cell chromatin state analysis with Signac
- PMID: 34725479
- PMCID: PMC9255697
- DOI: 10.1038/s41592-021-01282-5
Single-cell chromatin state analysis with Signac
Erratum in
-
Author Correction: Single-cell chromatin state analysis with Signac.Nat Methods. 2022 Feb;19(2):257. doi: 10.1038/s41592-022-01393-7. Nat Methods. 2022. PMID: 34997233 No abstract available.
Abstract
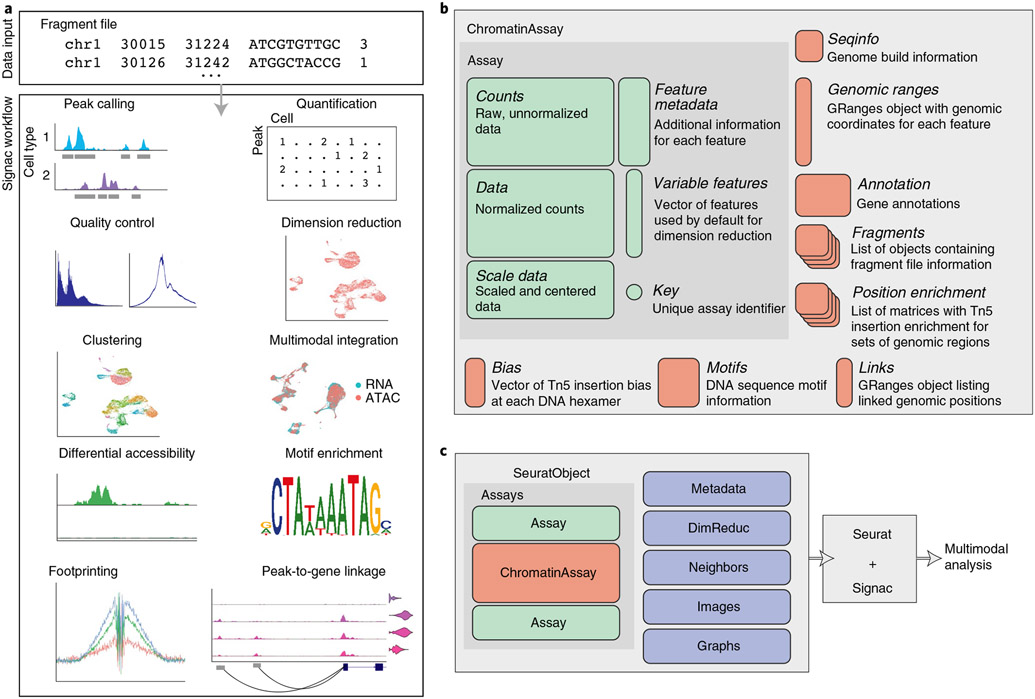
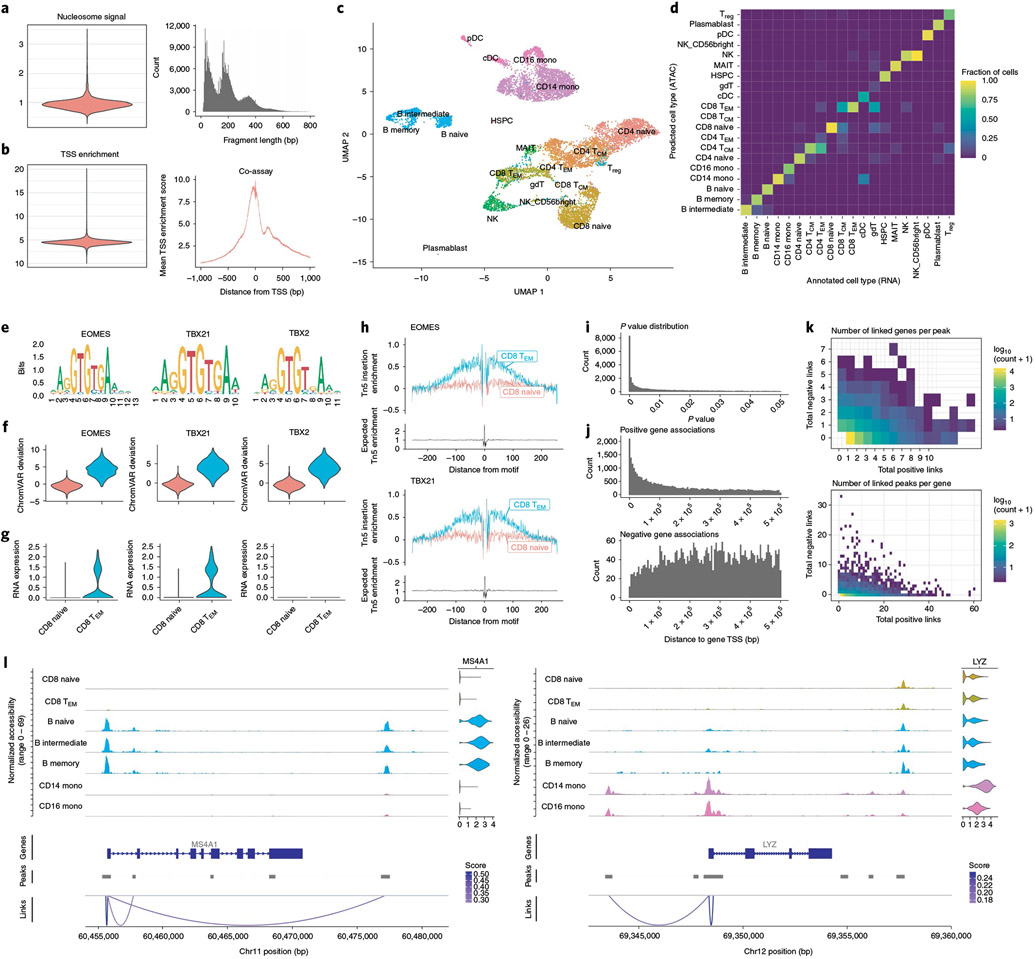
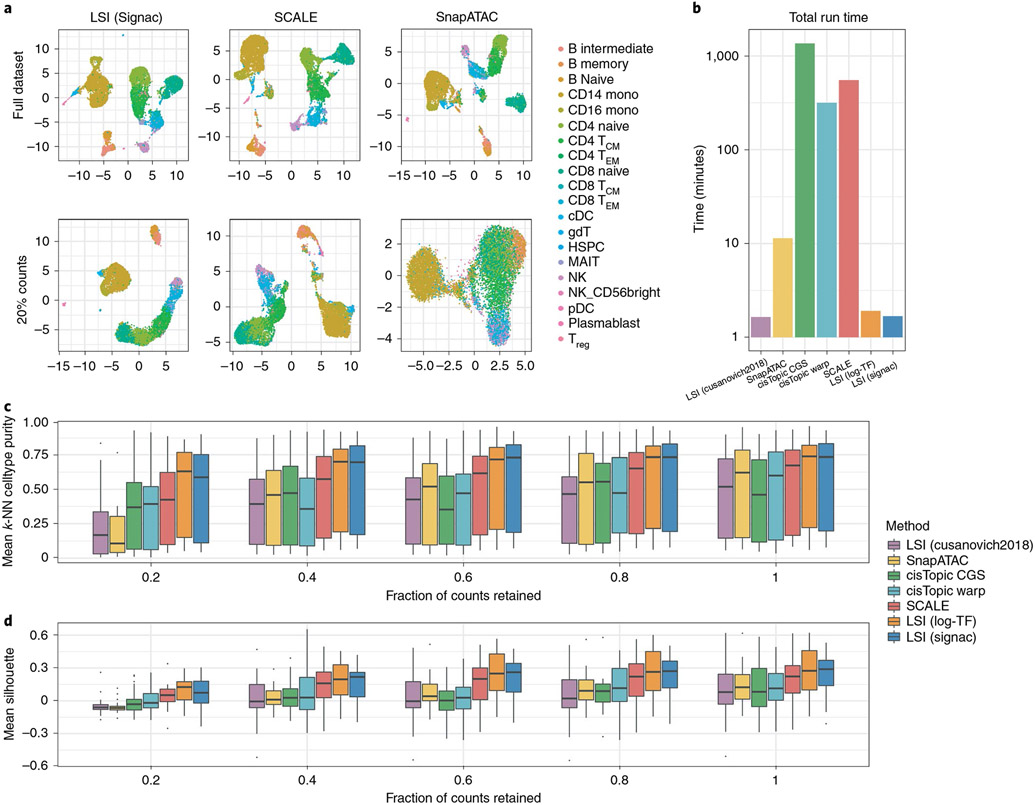
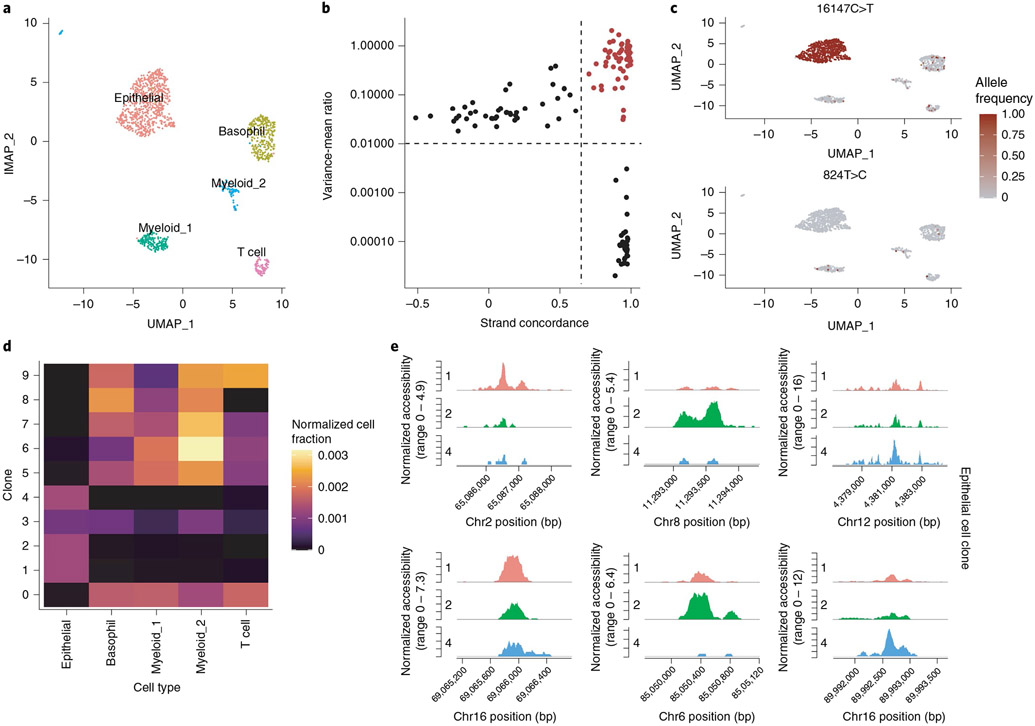
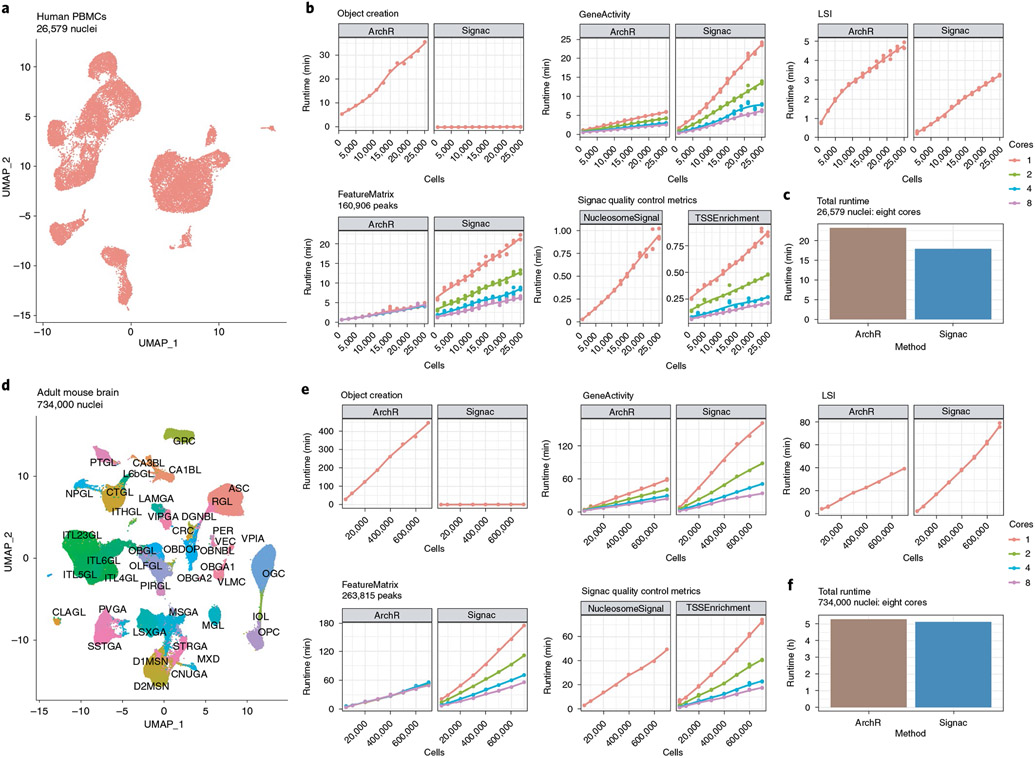
The recent development of experimental methods for measuring chromatin state at single-cell resolution has created a need for computational tools capable of analyzing these datasets. Here we developed Signac, a comprehensive toolkit for the analysis of single-cell chromatin data. Signac enables an end-to-end analysis of single-cell chromatin data, including peak calling, quantification, quality control, dimension reduction, clustering, integration with single-cell gene expression datasets, DNA motif analysis and interactive visualization. Through its seamless compatibility with the Seurat package, Signac facilitates the analysis of diverse multimodal single-cell chromatin data, including datasets that co-assay DNA accessibility with gene expression, protein abundance and mitochondrial genotype. We demonstrate scaling of the Signac framework to analyze datasets containing over 700,000 cells.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
-
- Ai S et al. Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol 21, 1164–1172 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
