

Nucleotide excision repair leaves a mark on chromatin: DNA damage detection in nucleosomes
- PMID: 34731255
- PMCID: PMC8629891
- DOI: 10.1007/s00018-021-03984-7
Nucleotide excision repair leaves a mark on chromatin: DNA damage detection in nucleosomes
Abstract
Global genome nucleotide excision repair (GG-NER) eliminates a broad spectrum of DNA lesions from genomic DNA. Genomic DNA is tightly wrapped around histones creating a barrier for DNA repair proteins to access DNA lesions buried in nucleosomal DNA. The DNA-damage sensors XPC and DDB2 recognize DNA lesions in nucleosomal DNA and initiate repair. The emerging view is that a tight interplay between XPC and DDB2 is regulated by post-translational modifications on the damage sensors themselves as well as on chromatin containing DNA lesions. The choreography between XPC and DDB2, their interconnection with post-translational modifications such as ubiquitylation, SUMOylation, methylation, poly(ADP-ribos)ylation, acetylation, and the functional links with chromatin remodelling activities regulate not only the initial recognition of DNA lesions in nucleosomes, but also the downstream recruitment and necessary displacement of GG-NER factors as repair progresses. In this review, we highlight how nucleotide excision repair leaves a mark on chromatin to enable DNA damage detection in nucleosomes.
Keywords: Chromatin; DDB2; Nucleotide excision repair; PTM; Post-translational modification; XPC.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
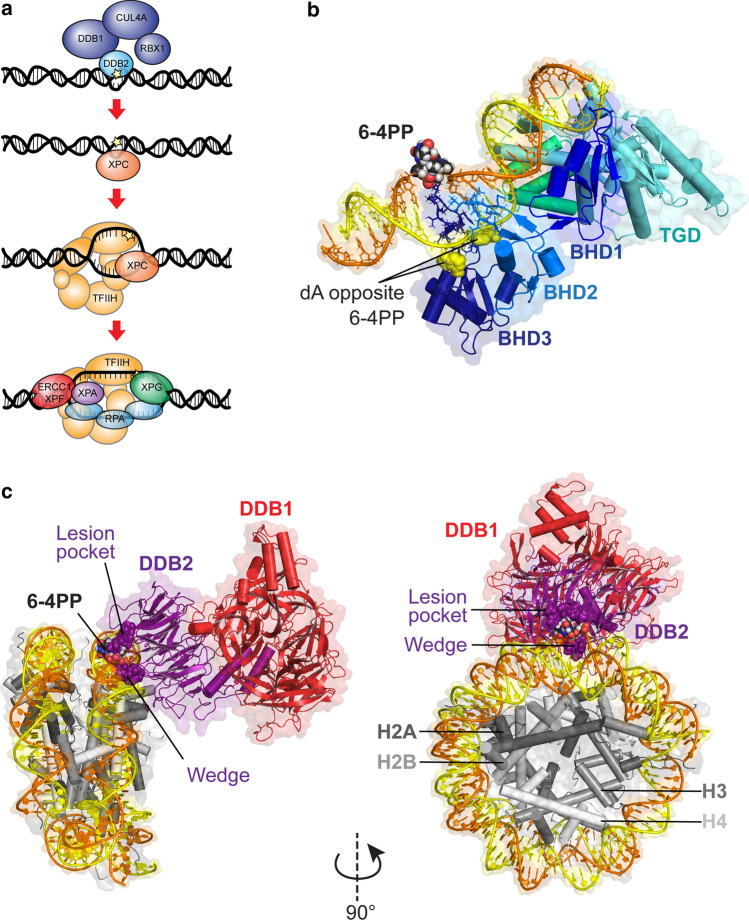
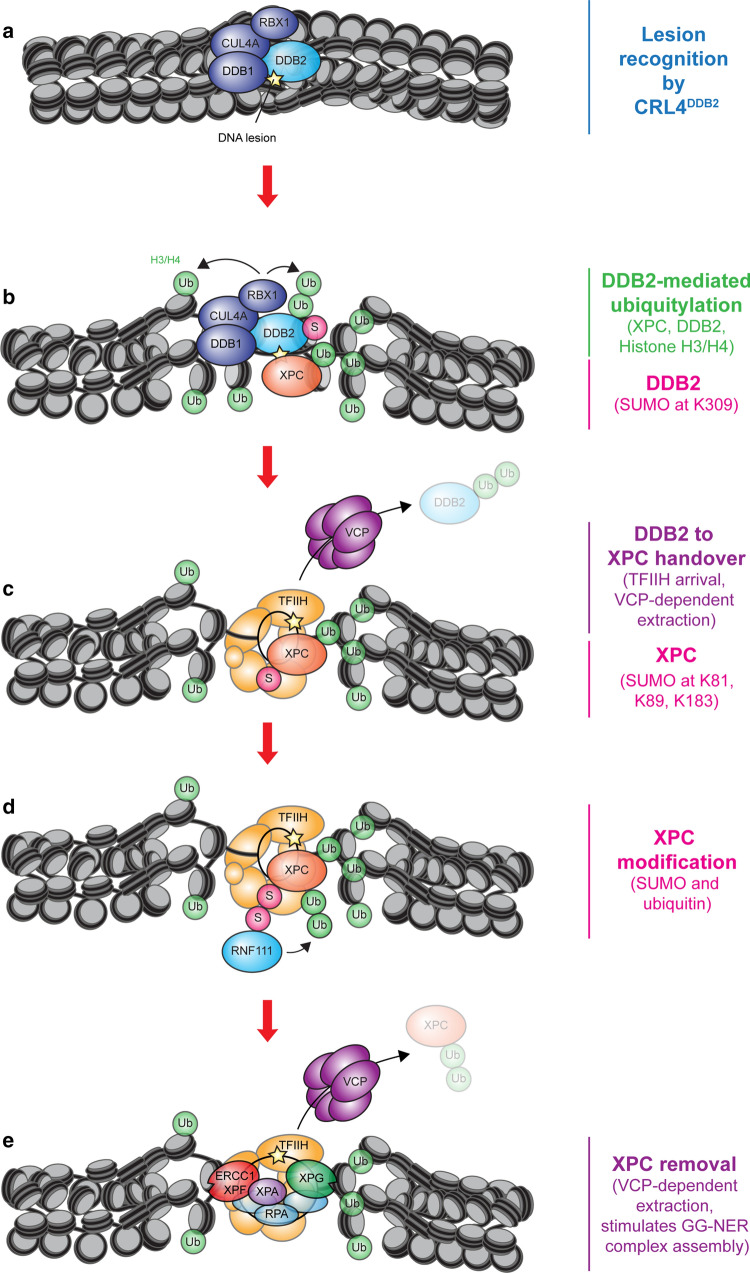
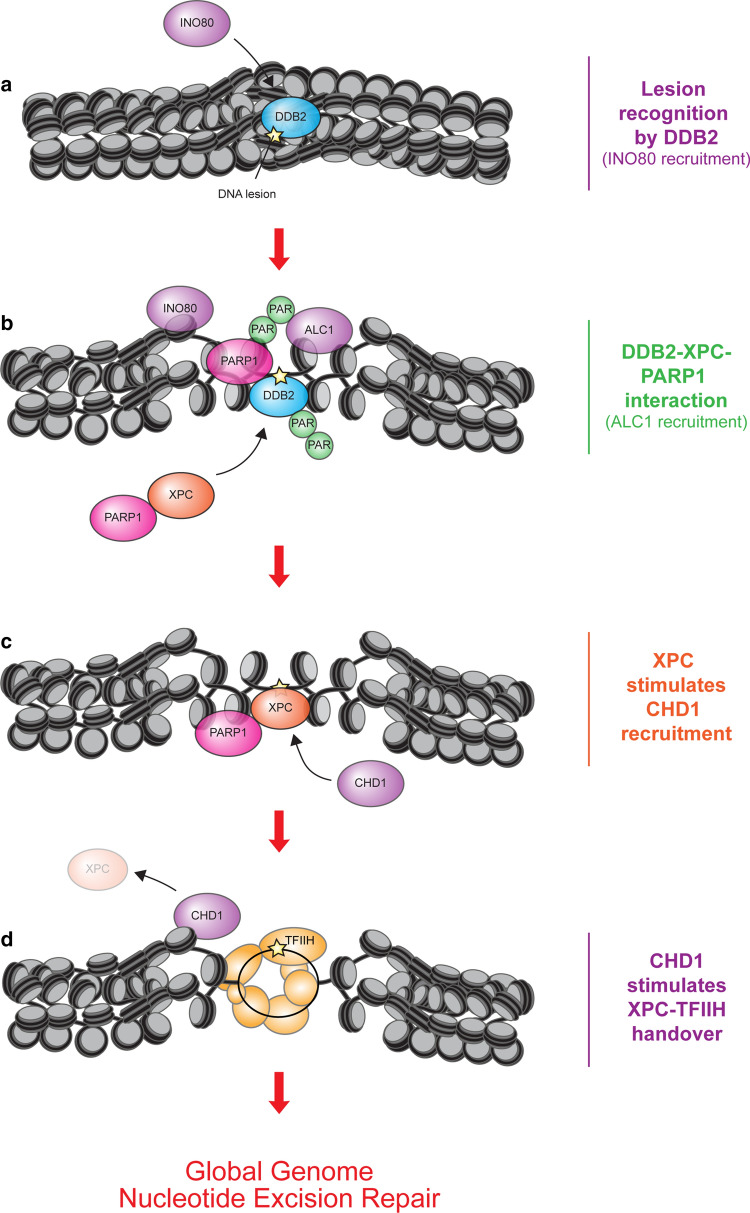
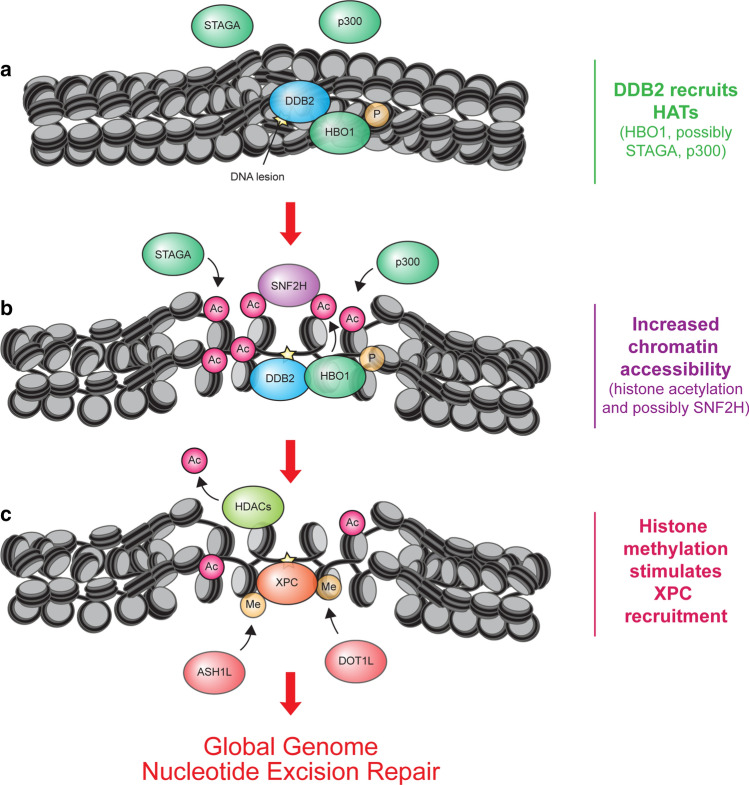
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
