

Use of whole genome sequencing to determine genetic basis of suspected mitochondrial disorders: cohort study
- PMID: 34732400
- PMCID: PMC8565085
- DOI: 10.1136/bmj-2021-066288
Use of whole genome sequencing to determine genetic basis of suspected mitochondrial disorders: cohort study
Abstract
Objective: To determine whether whole genome sequencing can be used to define the molecular basis of suspected mitochondrial disease.
Design: Cohort study.
Setting: National Health Service, England, including secondary and tertiary care.
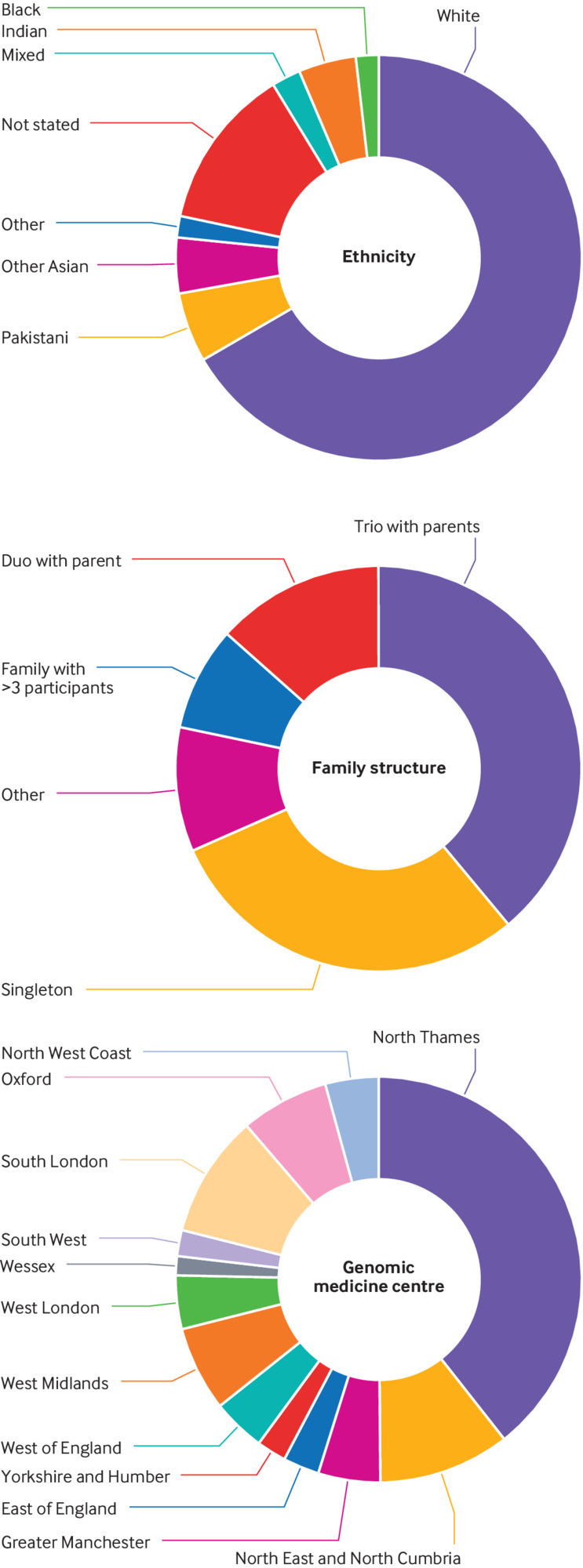
Participants: 345 patients with suspected mitochondrial disorders recruited to the 100 000 Genomes Project in England between 2015 and 2018.
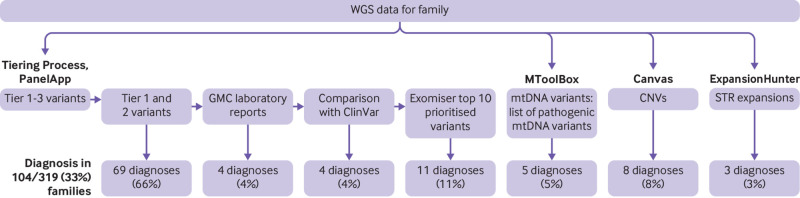
Intervention: Short read whole genome sequencing was performed. Nuclear variants were prioritised on the basis of gene panels chosen according to phenotypes, ClinVar pathogenic/likely pathogenic variants, and the top 10 prioritised variants from Exomiser. Mitochondrial DNA variants were called using an in-house pipeline and compared with a list of pathogenic variants. Copy number variants and short tandem repeats for 13 neurological disorders were also analysed. American College of Medical Genetics guidelines were followed for classification of variants.
Main outcome measure: Definite or probable genetic diagnosis.
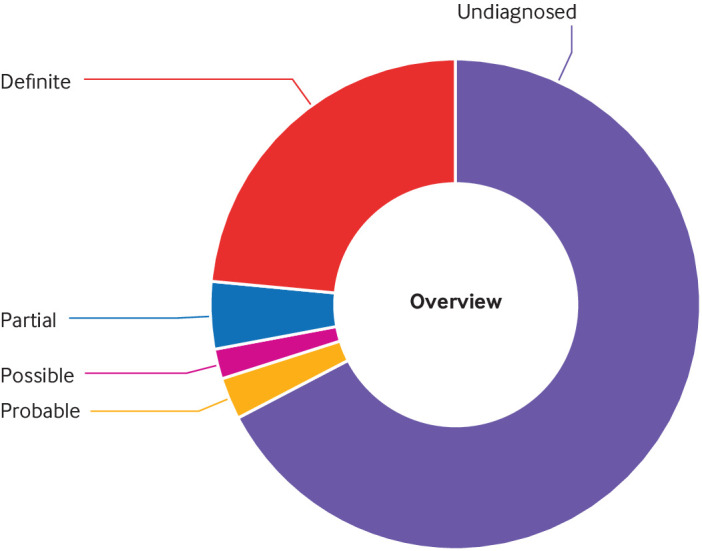
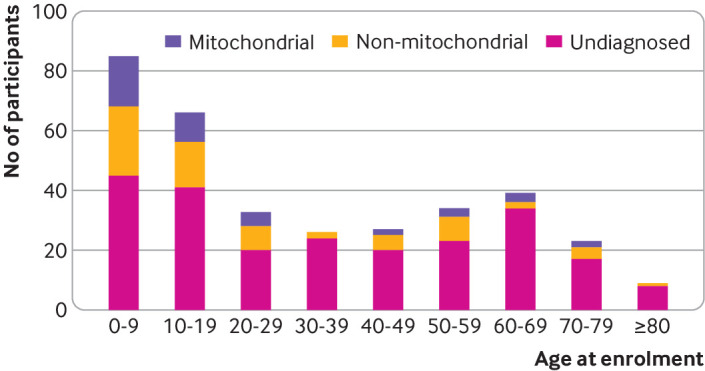
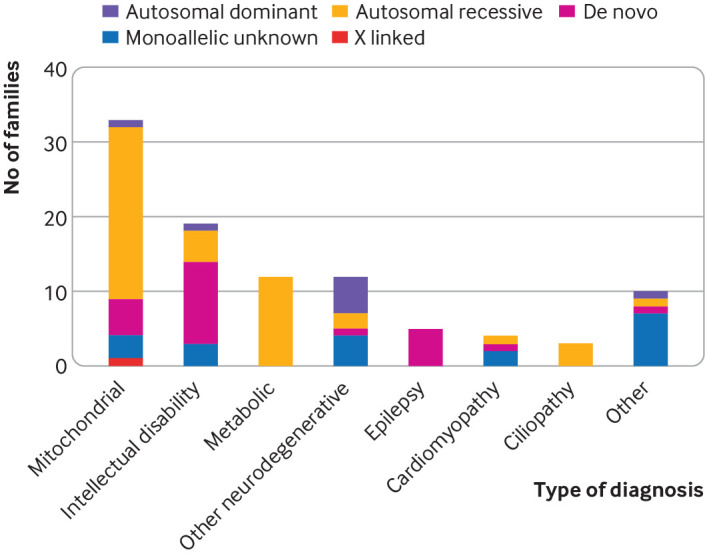
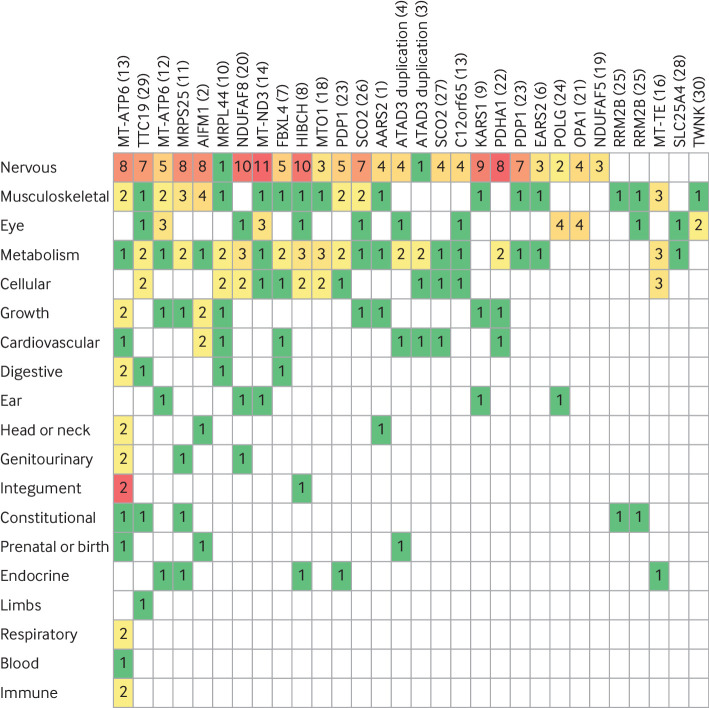
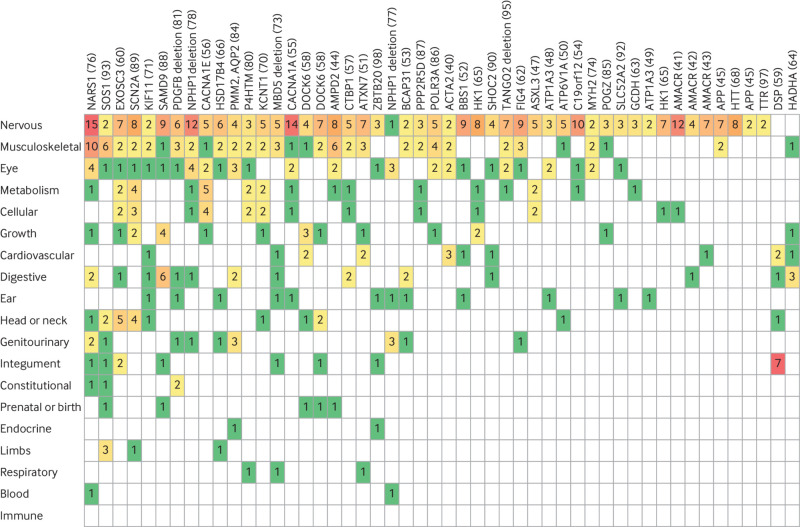
Results: A definite or probable genetic diagnosis was identified in 98/319 (31%) families, with an additional 6 (2%) possible diagnoses. Fourteen of the diagnoses (4% of the 319 families) explained only part of the clinical features. A total of 95 different genes were implicated. Of 104 families given a diagnosis, 39 (38%) had a mitochondrial diagnosis and 65 (63%) had a non-mitochondrial diagnosis.
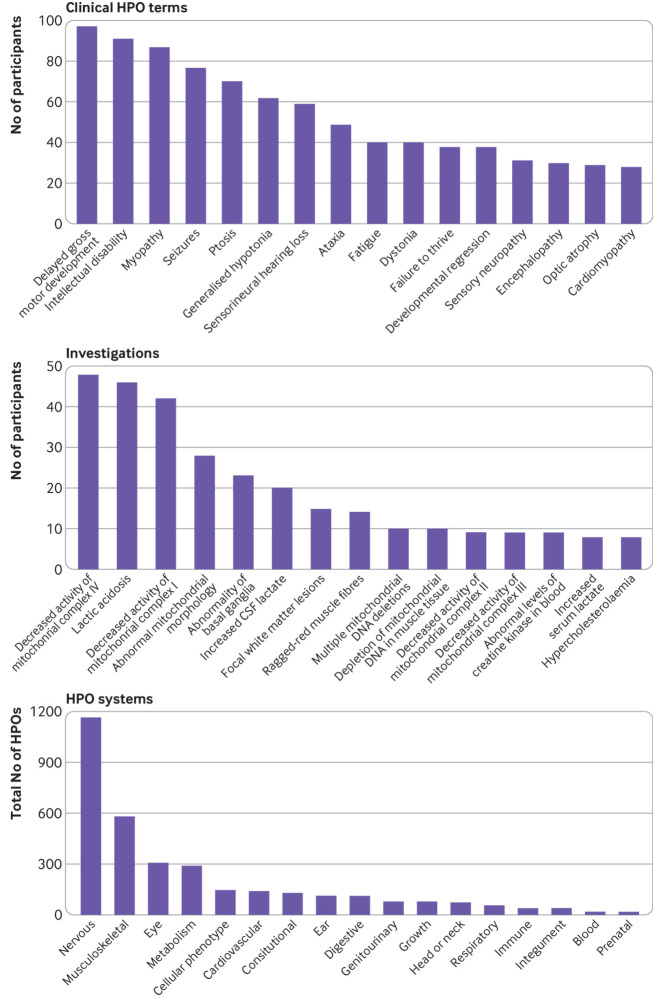
Conclusion: Whole genome sequencing is a useful diagnostic test in patients with suspected mitochondrial disorders, yielding a diagnosis in a further 31% after exclusion of common causes. Most diagnoses were non-mitochondrial disorders and included developmental disorders with intellectual disability, epileptic encephalopathies, other metabolic disorders, cardiomyopathies, and leukodystrophies. These would have been missed if a targeted approach was taken, and some have specific treatments.
© Author(s) (or their employer(s)) 2019. Re-use permitted under CC BY. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing Interests: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/disclosure-of-interest/ and declare: support for the submitted work as detailed above; no financial relationships with any organisations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- G1000848/MRC_/Medical Research Council/United Kingdom
- MC_PC_14089/MRC_/Medical Research Council/United Kingdom
- MR/S035699/1/MRC_/Medical Research Council/United Kingdom
- MC_EX_MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- MR/N025431/1/MRC_/Medical Research Council/United Kingdom
- MR/S002065/1/MRC_/Medical Research Council/United Kingdom
- MR/V009346/1/MRC_/Medical Research Council/United Kingdom
- MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- MR/N025431/2/MRC_/Medical Research Council/United Kingdom
- MC_UU_00015/9/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- MR/S006753/1/MRC_/Medical Research Council/United Kingdom
- MR/N010035/1/MRC_/Medical Research Council/United Kingdom
- MR/S005021/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical