

Monitoring the Microevolution of Salmonella enterica in Healthy Dairy Cattle Populations at the Individual Farm Level Using Whole-Genome Sequencing
- PMID: 34733267
- PMCID: PMC8558520
- DOI: 10.3389/fmicb.2021.763669
Monitoring the Microevolution of Salmonella enterica in Healthy Dairy Cattle Populations at the Individual Farm Level Using Whole-Genome Sequencing
Abstract
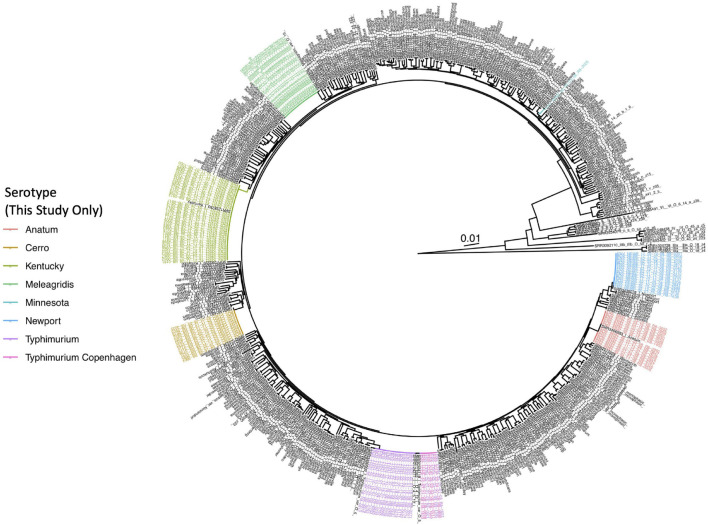
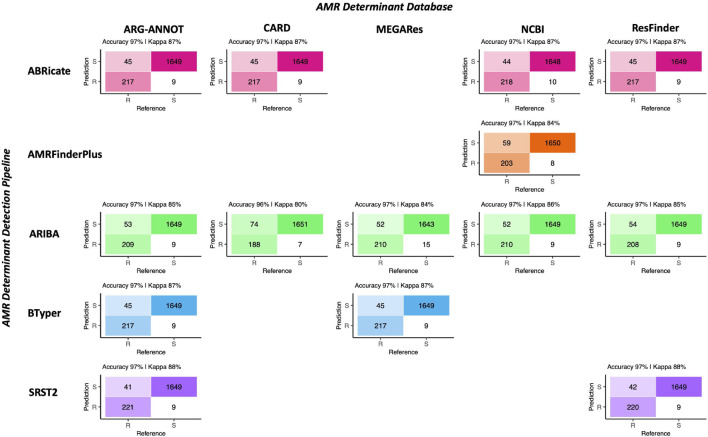
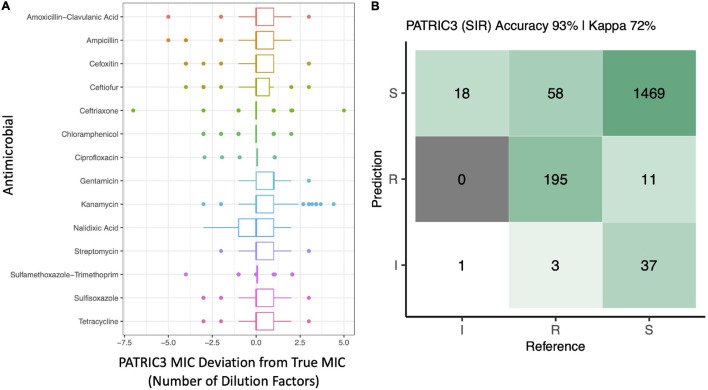
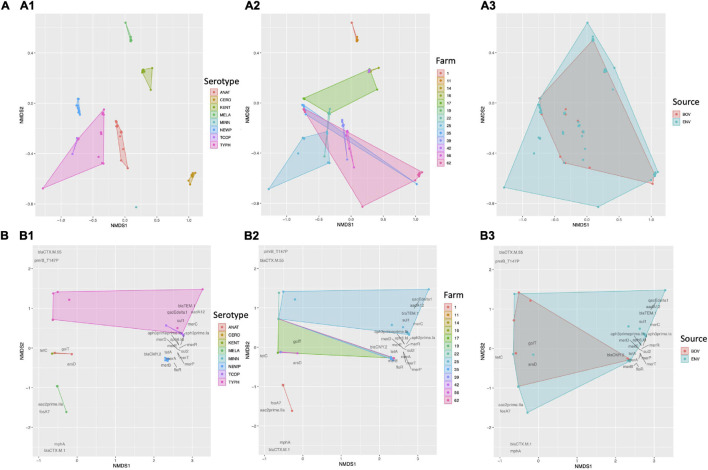
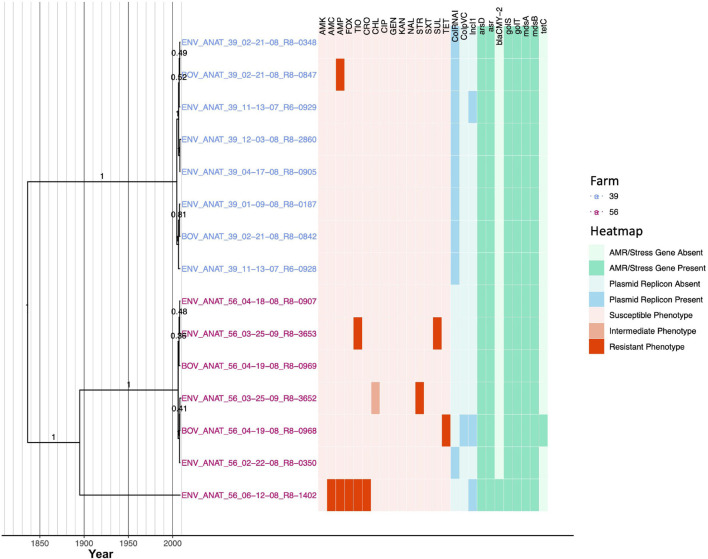
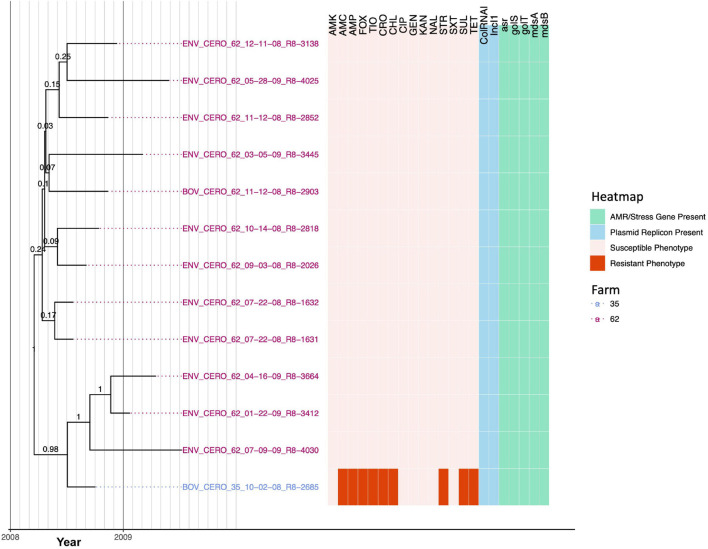
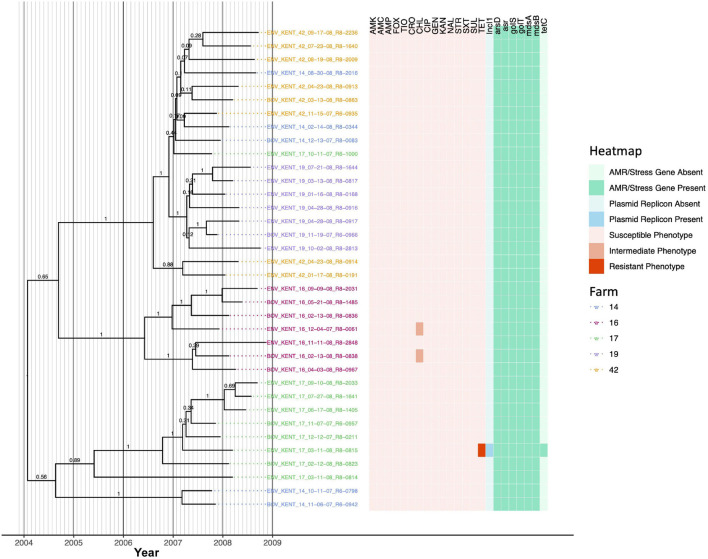
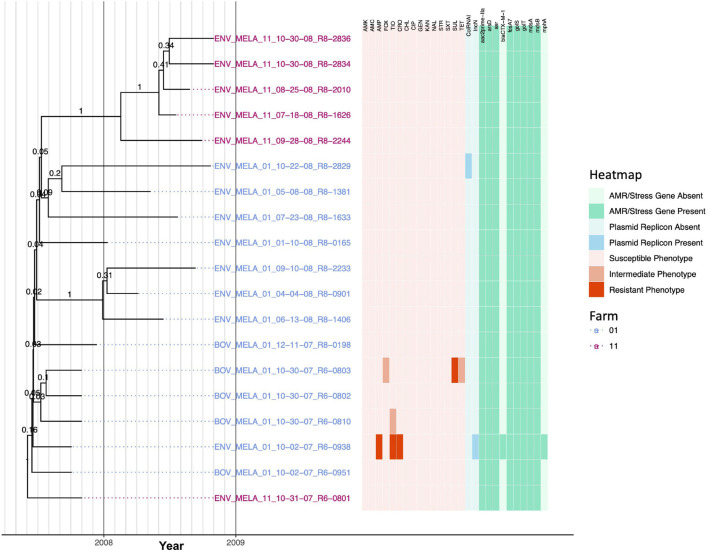
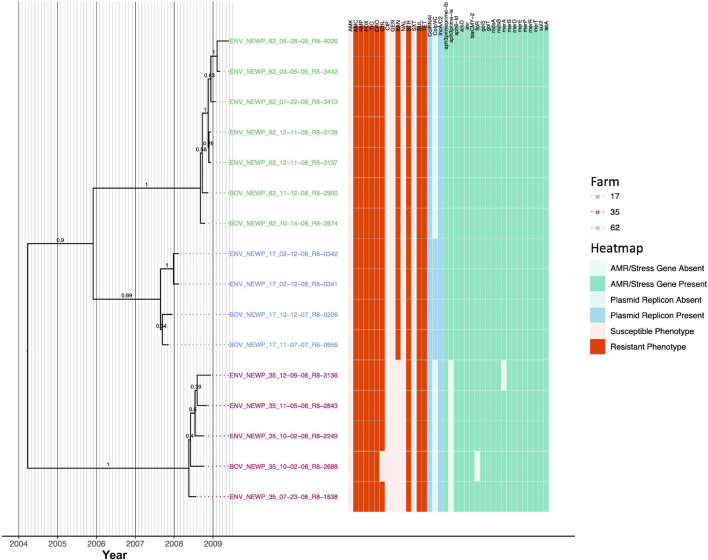
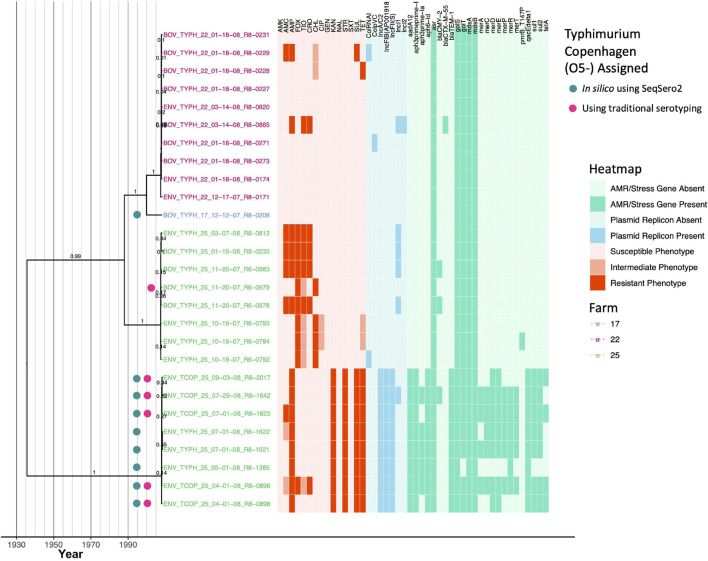
Livestock represent a possible reservoir for facilitating the transmission of the zoonotic foodborne pathogen Salmonella enterica to humans; there is also concern that strains can acquire resistance to antimicrobials in the farm environment. Here, whole-genome sequencing (WGS) was used to characterize Salmonella strains (n = 128) isolated from healthy dairy cattle and their associated environments on 13 New York State farms to assess the diversity and microevolution of this important pathogen at the level of the individual herd. Additionally, the accuracy and concordance of multiple in silico tools are assessed, including: (i) two in silico serotyping tools, (ii) combinations of five antimicrobial resistance (AMR) determinant detection tools and one to five AMR determinant databases, and (iii) one antimicrobial minimum inhibitory concentration (MIC) prediction tool. For the isolates sequenced here, in silico serotyping methods outperformed traditional serotyping and resolved all un-typable and/or ambiguous serotype assignments. Serotypes assigned in silico showed greater congruency with the Salmonella whole-genome phylogeny than traditional serotype assignments, and in silico methods showed high concordance (99% agreement). In silico AMR determinant detection methods additionally showed a high degree of concordance, regardless of the pipeline or database used (≥98% agreement among susceptible/resistant assignments for all pipeline/database combinations). For AMR detection methods that relied exclusively on nucleotide BLAST, accuracy could be maximized by using a range of minimum nucleotide identity and coverage thresholds, with thresholds of 75% nucleotide identity and 50-60% coverage adequate for most pipeline/database combinations. In silico characterization of the microevolution and AMR dynamics of each of six serotype groups (S. Anatum, Cerro, Kentucky, Meleagridis, Newport, Typhimurium/Typhimurium variant Copenhagen) revealed that some lineages were strongly associated with individual farms, while others were distributed across multiple farms. Numerous AMR determinant acquisition and loss events were identified, including the recent acquisition of cephalosporin resistance-conferring bla CMY- and bla CTX-M-type beta-lactamases. The results presented here provide high-resolution insight into the temporal dynamics of AMR Salmonella at the scale of the individual farm and highlight both the strengths and limitations of WGS in tracking zoonotic pathogens and their associated AMR determinants at the livestock-human interface.
Keywords: Salmonella; antimicrobial resistance; dairy cattle; evolution; livestock; serotyping; whole-genome sequencing.
Copyright © 2021 Carroll, Buehler, Gaballa, Siler, Cummings, Cheng and Wiedmann.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Agren E. C., Wahlstrom H., Vesterlund-Carlson C., Lahti E., Melin L., Soderlund R. (2016). Comparison of whole genome sequencing typing results and epidemiological contact information from outbreaks of Salmonella Dublin in Swedish cattle herds. Infect. Ecol. Epidemiol. 6:31782. 10.3402/iee.v6.31782 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials