

Chemotherapy induces canalization of cell state in childhood B-cell precursor acute lymphoblastic leukemia
- PMID: 34734190
- PMCID: PMC7611923
- DOI: 10.1038/s43018-021-00219-3
Chemotherapy induces canalization of cell state in childhood B-cell precursor acute lymphoblastic leukemia
Abstract
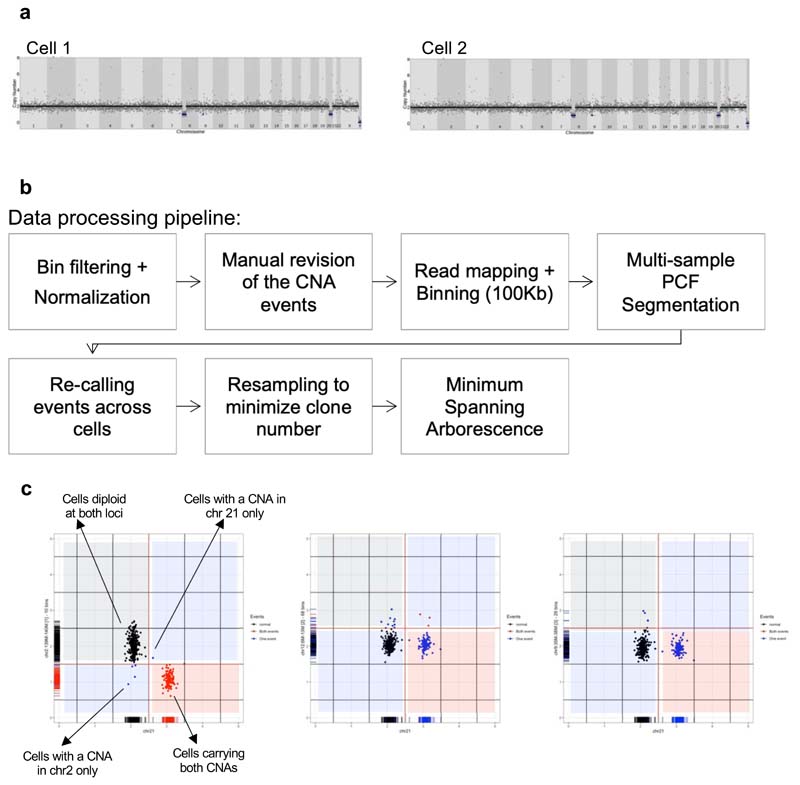
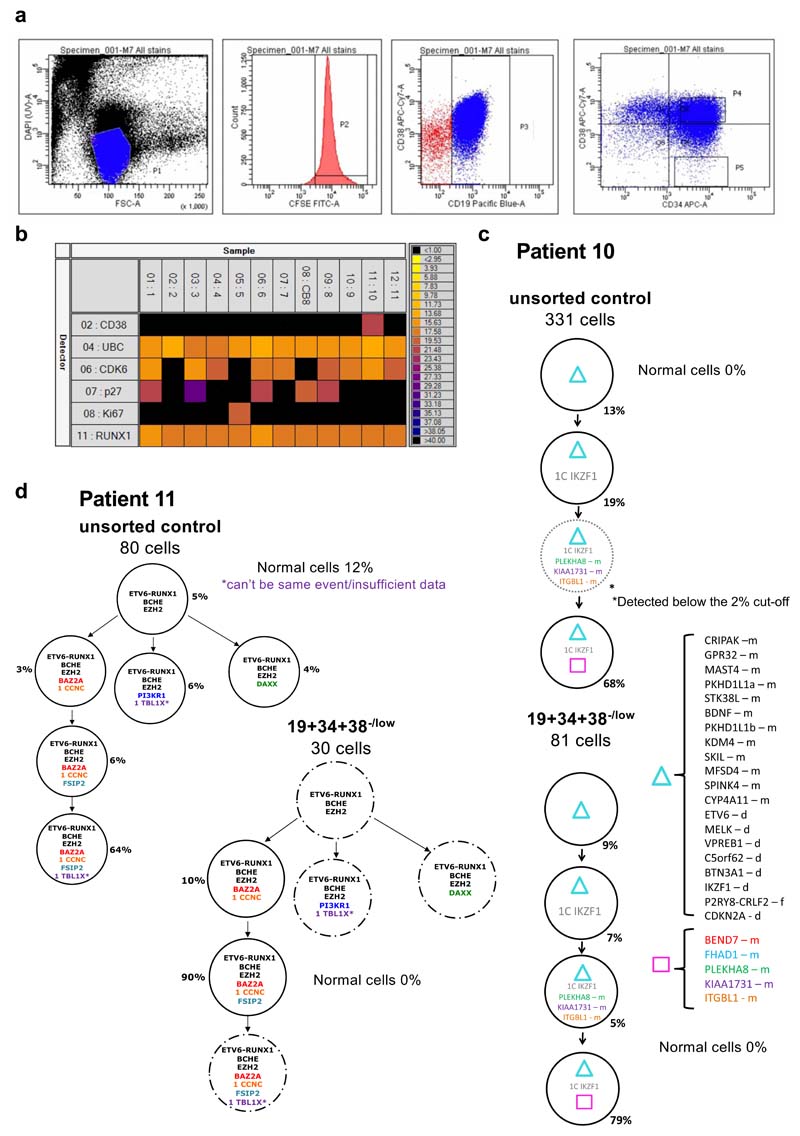
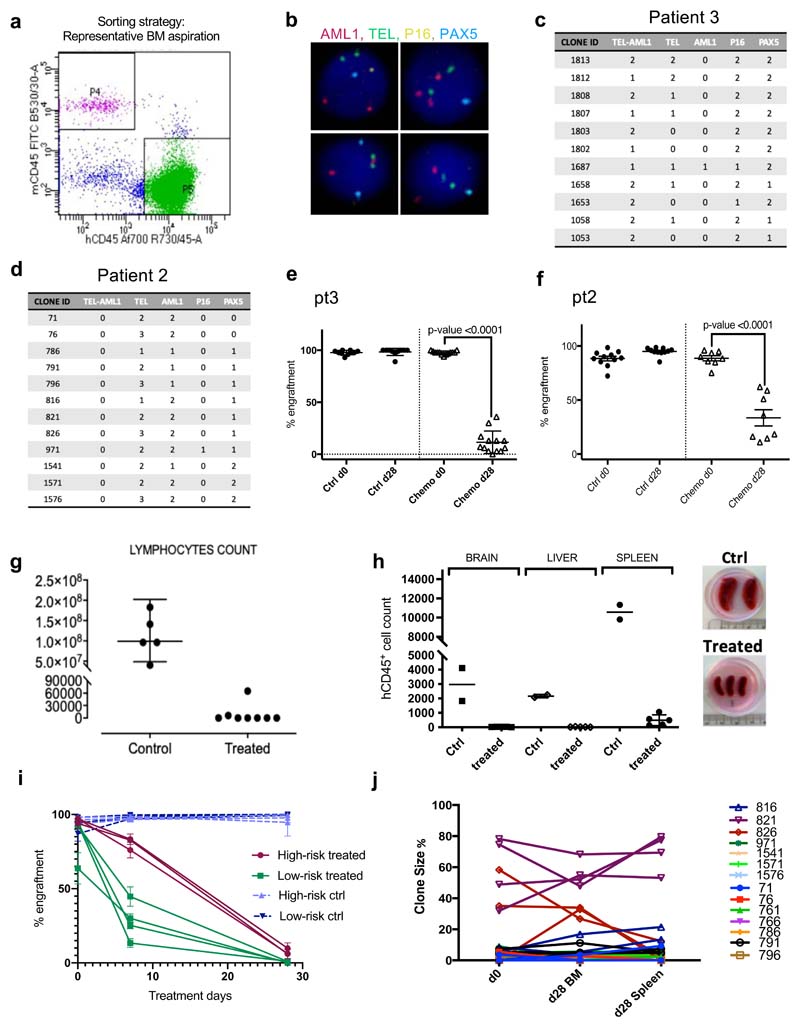
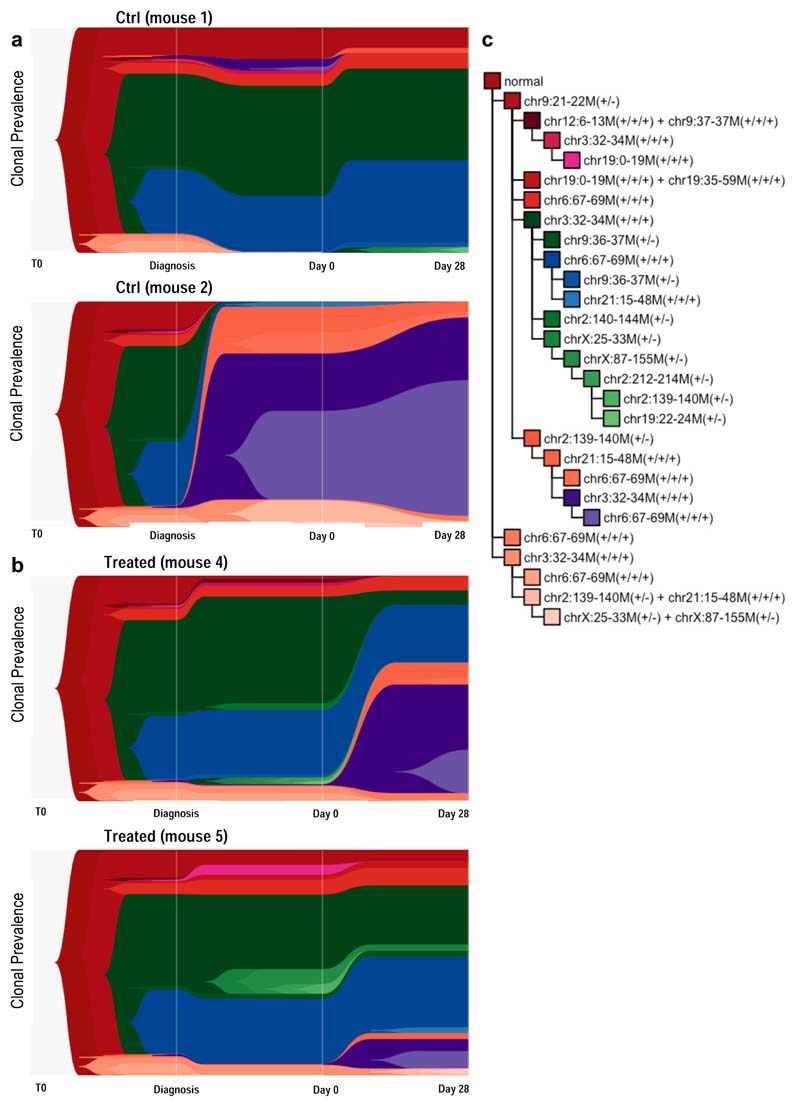
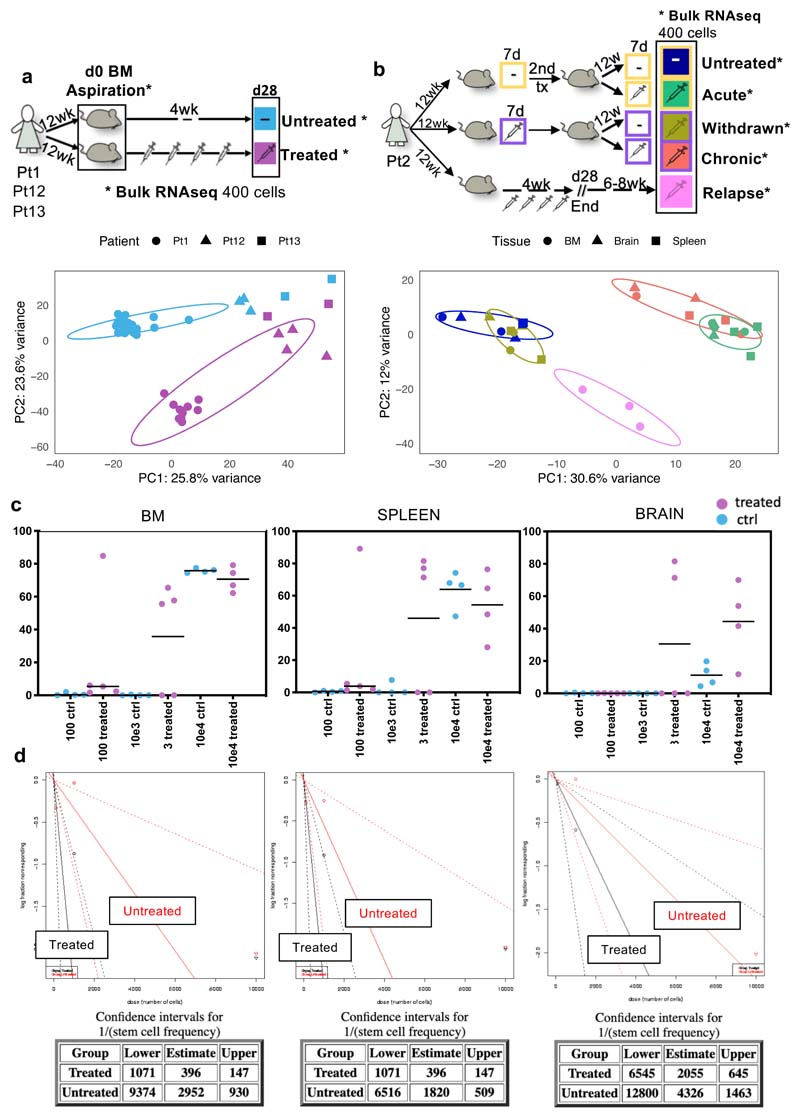
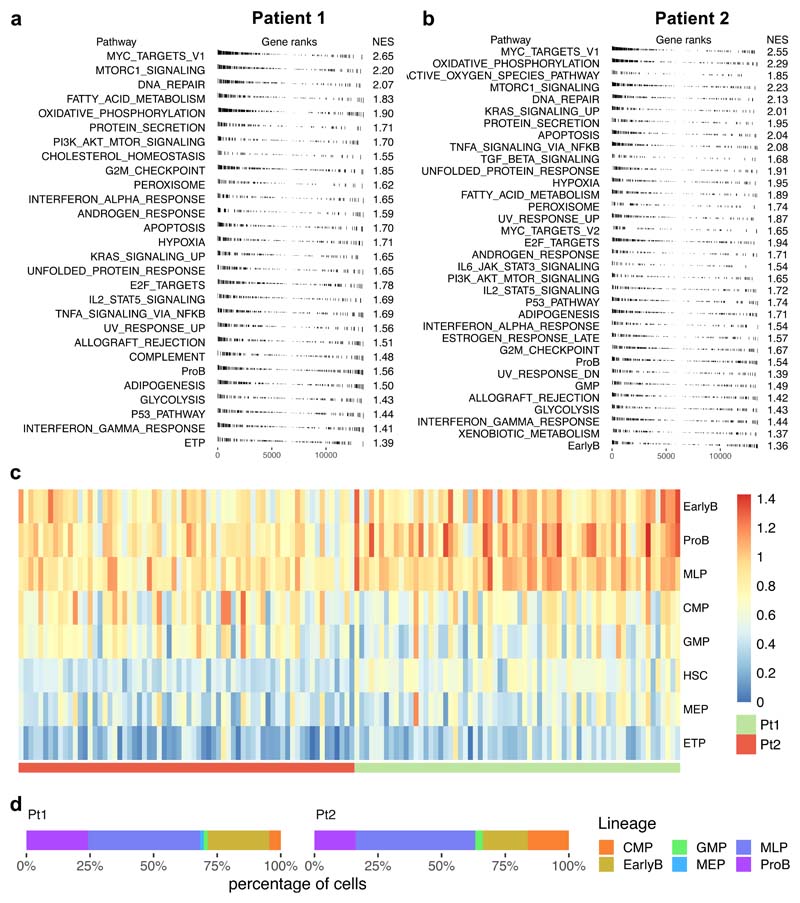
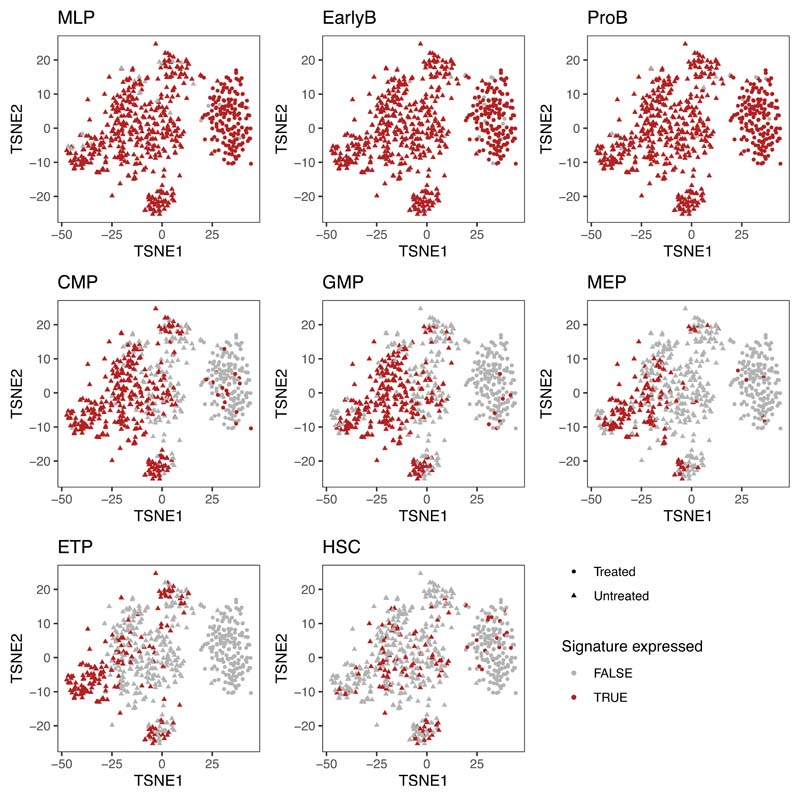
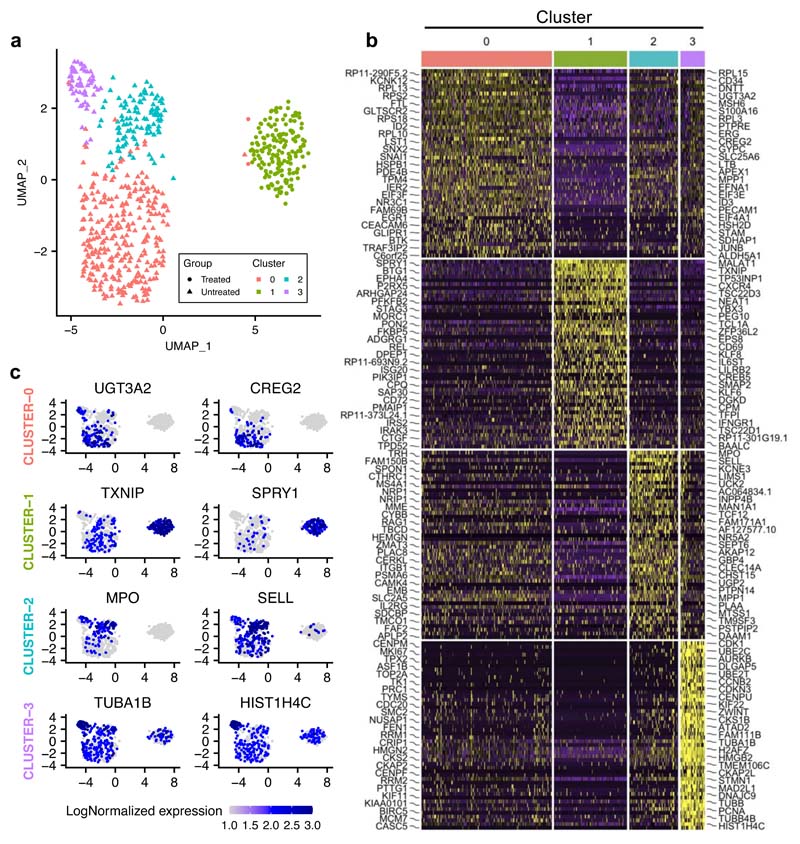
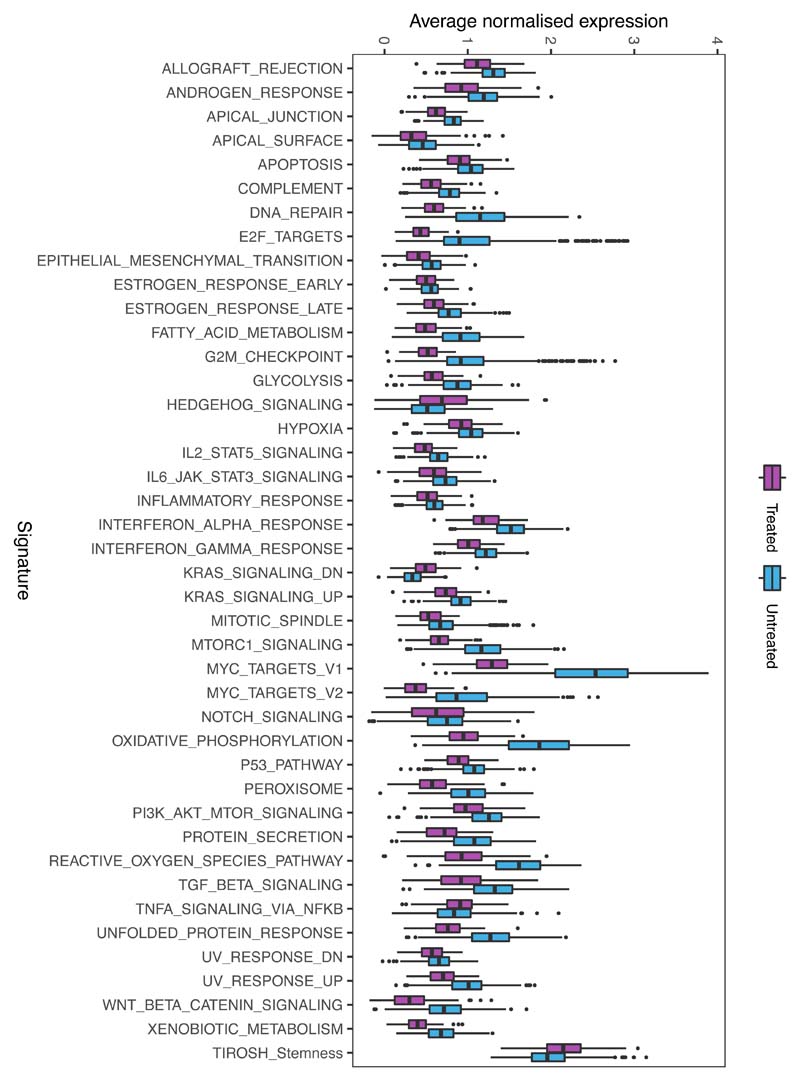
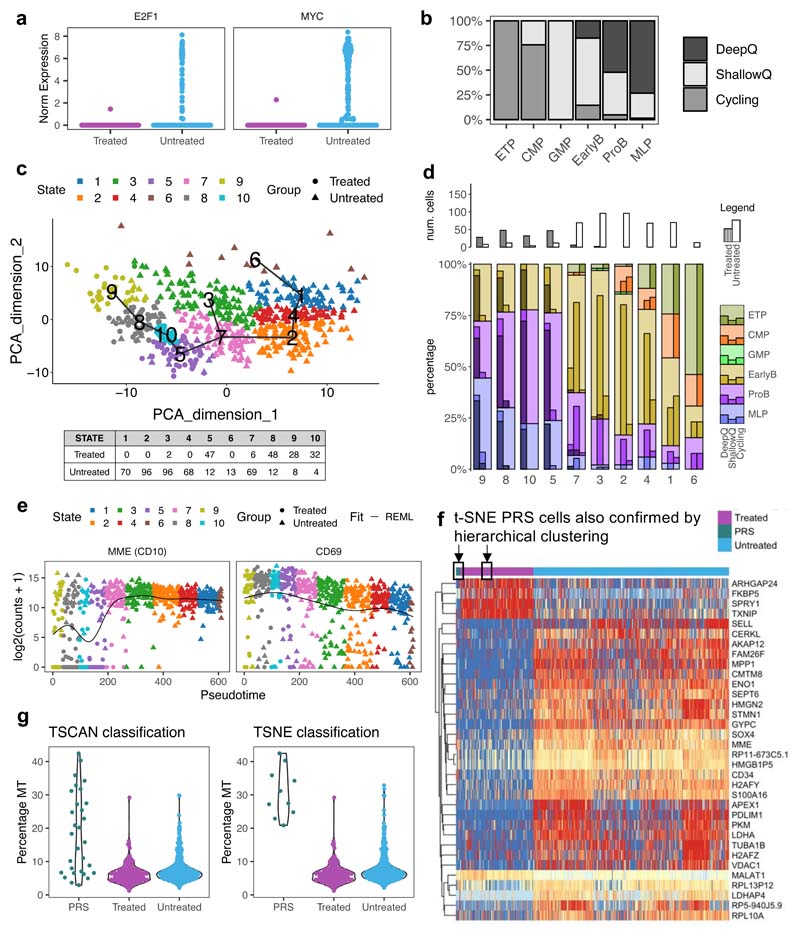
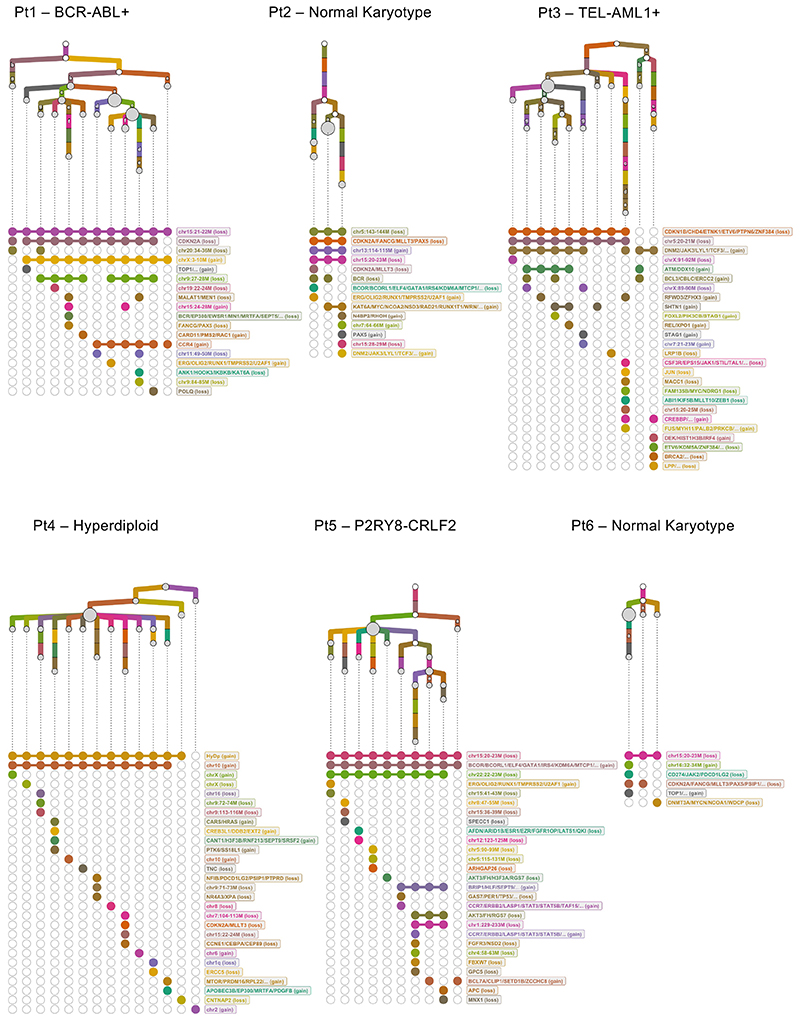
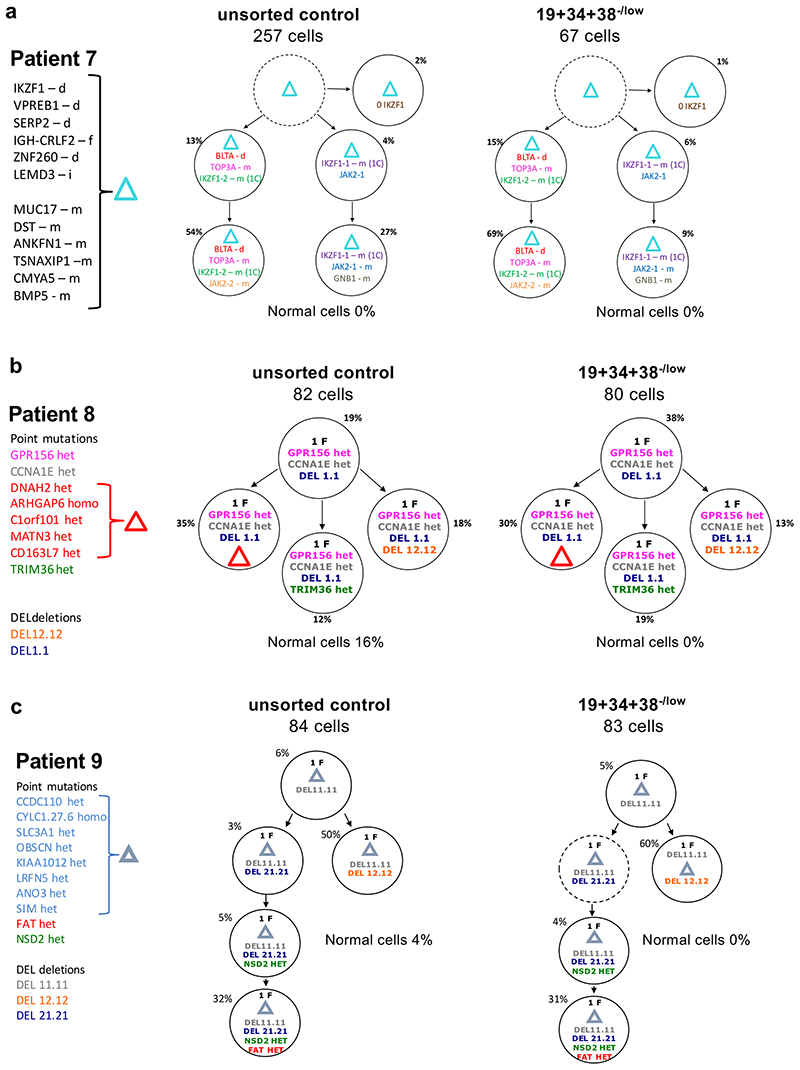
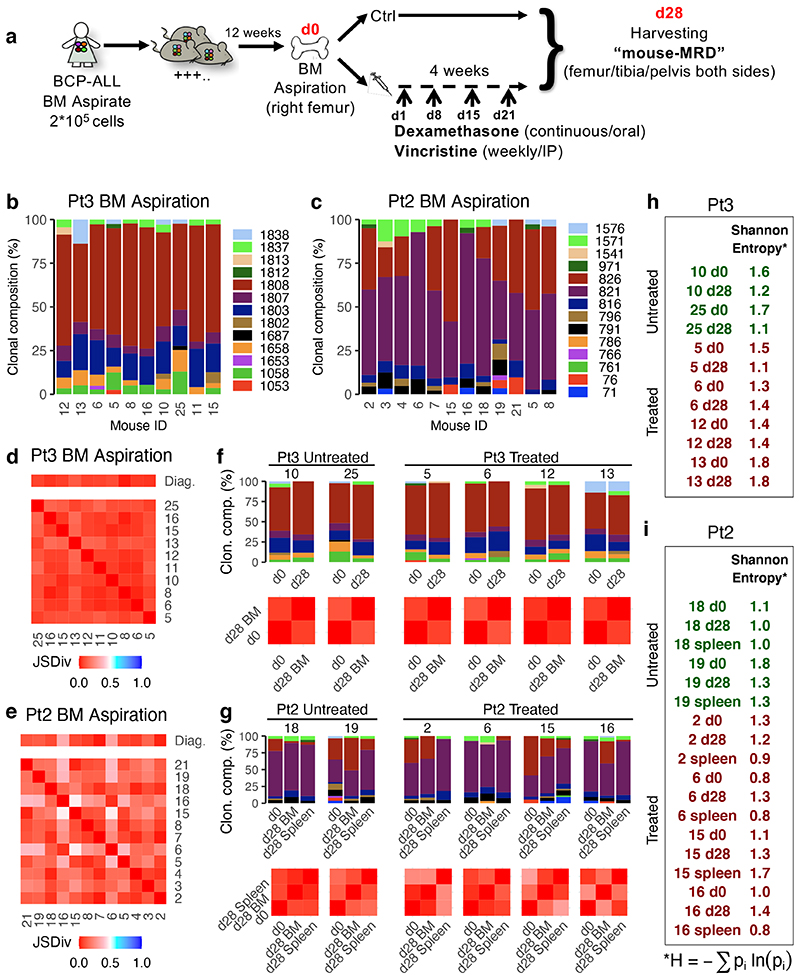
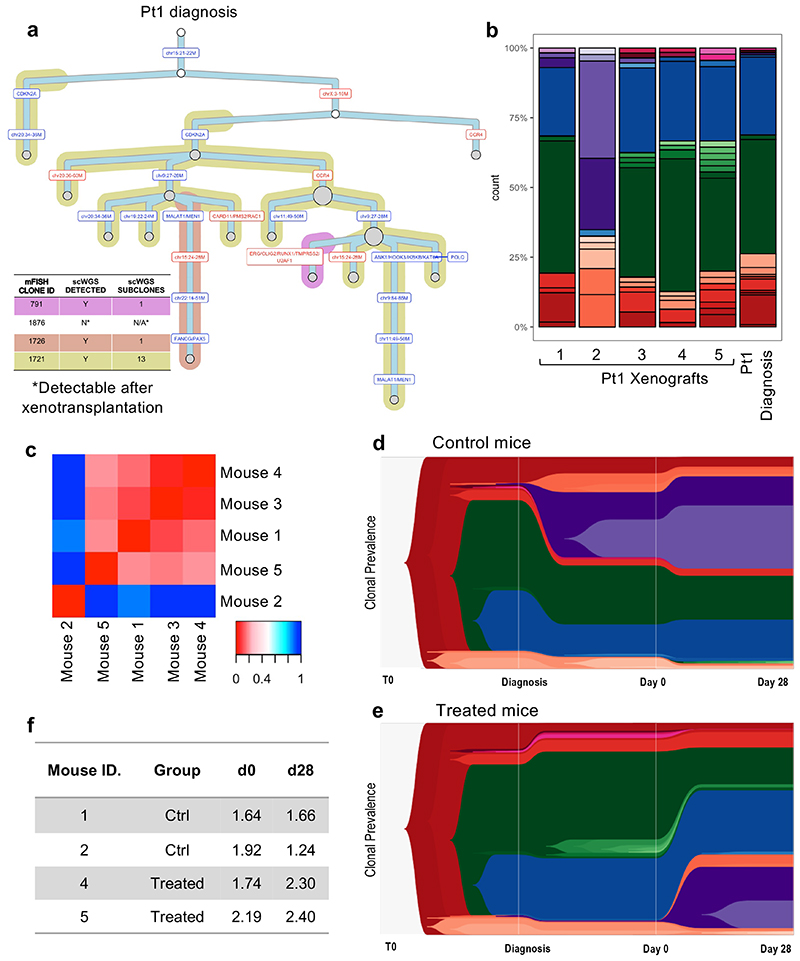
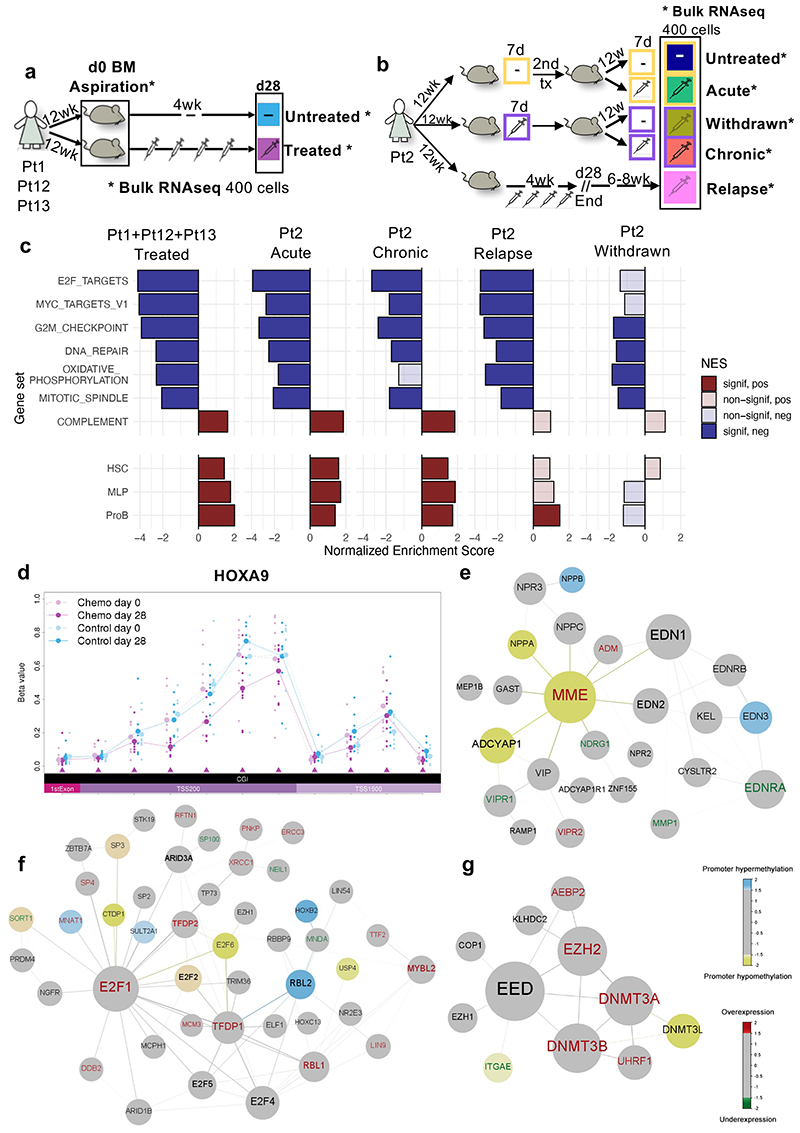
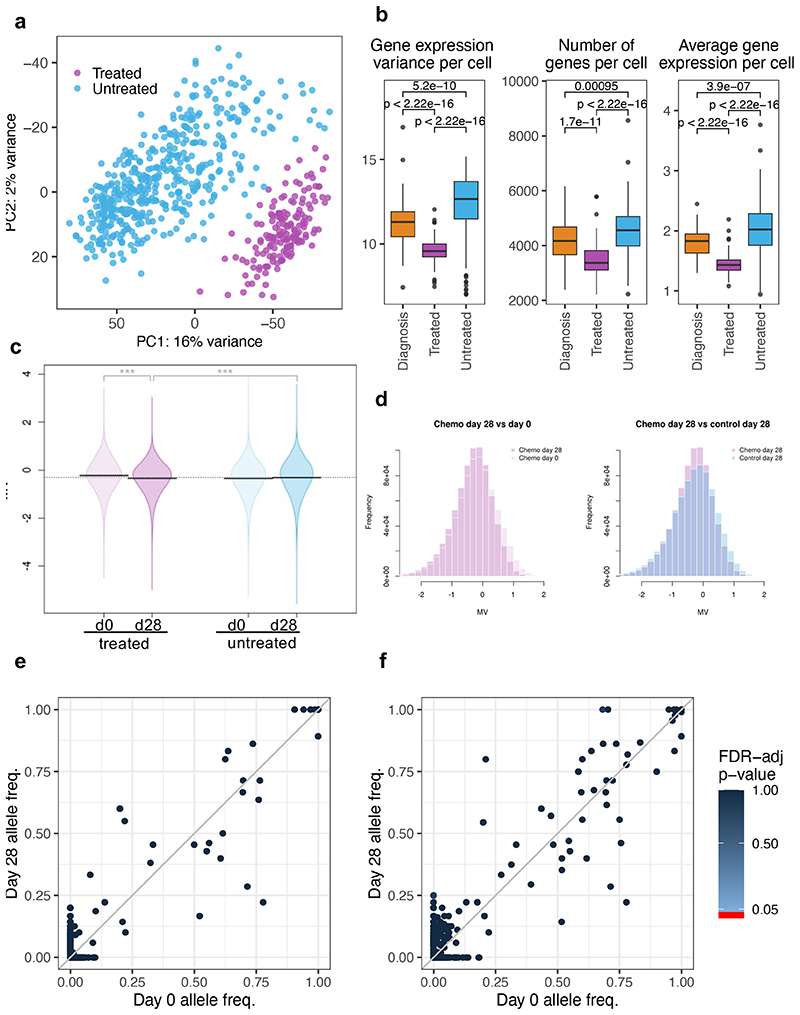
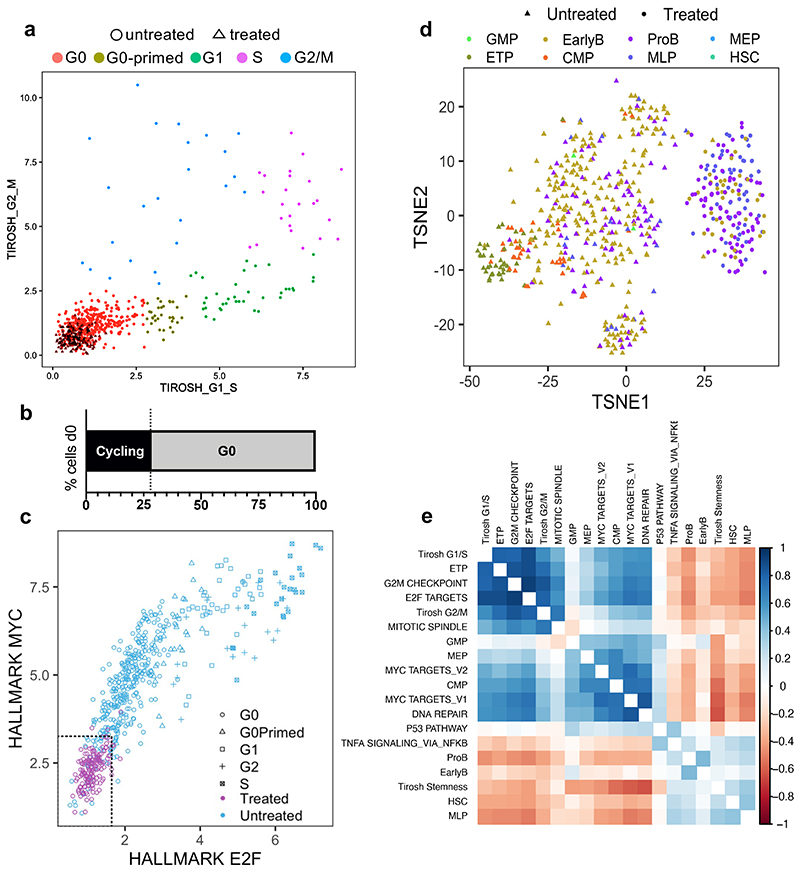
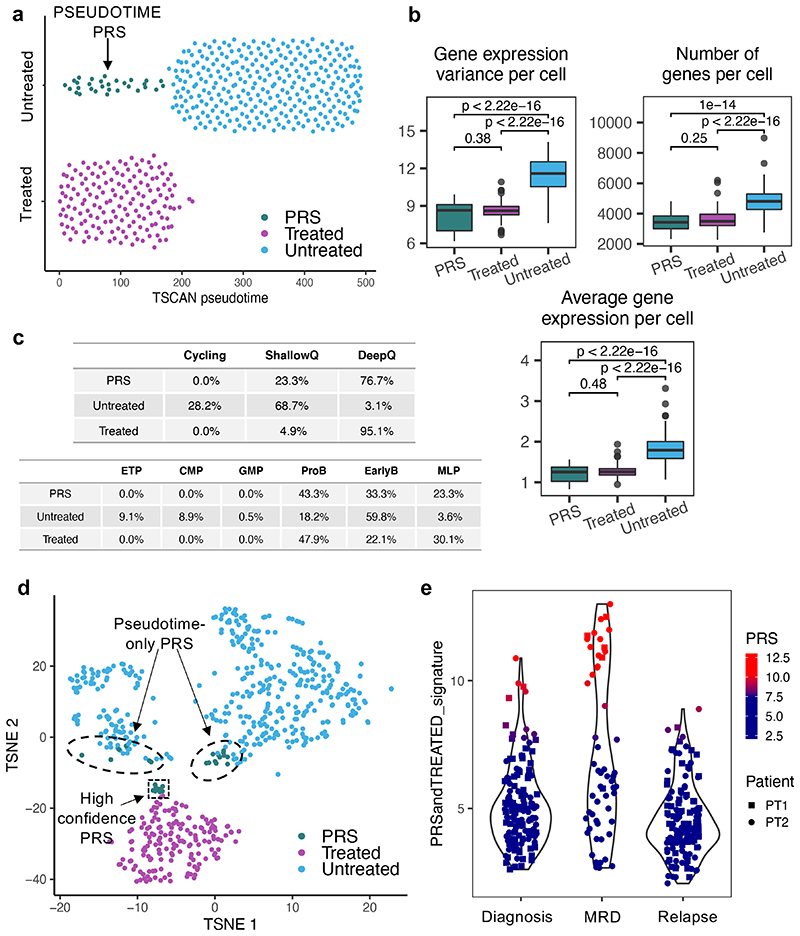
Comparison of intratumor genetic heterogeneity in cancer at diagnosis and relapse suggests that chemotherapy induces bottleneck selection of subclonal genotypes. However, evolutionary events subsequent to chemotherapy could also explain changes in clonal dominance seen at relapse. We, therefore, investigated the mechanisms of selection in childhood B-cell precursor acute lymphoblastic leukemia (BCP-ALL) during induction chemotherapy where maximal cytoreduction occurs. To distinguish stochastic versus deterministic events, individual leukemias were transplanted into multiple xenografts and chemotherapy administered. Analyses of the immediate post-treatment leukemic residuum at single-cell resolution revealed that chemotherapy has little impact on genetic heterogeneity. Rather, it acts on extensive, previously unappreciated, transcriptional and epigenetic heterogeneity in BCP-ALL, dramatically reducing the spectrum of cell states represented, leaving a genetically polyclonal but phenotypically uniform population with hallmark signatures relating to developmental stage, cell cycle and metabolism. Hence, canalization of cell state accounts for a significant component of bottleneck selection during induction chemotherapy.
Conflict of interest statement
Competing Interest M.L was an employee of Fluidigm Corporation at the time of the study.
Figures
References
-
- Almendro V, Marusyk A, Polyak K. Cellular Heterogeneity and Molecular Evolution in Cancer. Annu Rev Pathol Mech Dis. 2013;8:277–302. - PubMed
-
- Anderson K, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. - PubMed
-
- Jamal-Hanjani M, et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N Engl J Med. 2017;376:2109–2121. - PubMed
-
- McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–628. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
