

DIPG Harbors Alterations Targetable by MEK Inhibitors, with Acquired Resistance Mechanisms Overcome by Combinatorial Inhibition
- PMID: 34737188
- PMCID: PMC7612484
- DOI: 10.1158/2159-8290.CD-20-0930
DIPG Harbors Alterations Targetable by MEK Inhibitors, with Acquired Resistance Mechanisms Overcome by Combinatorial Inhibition
Abstract
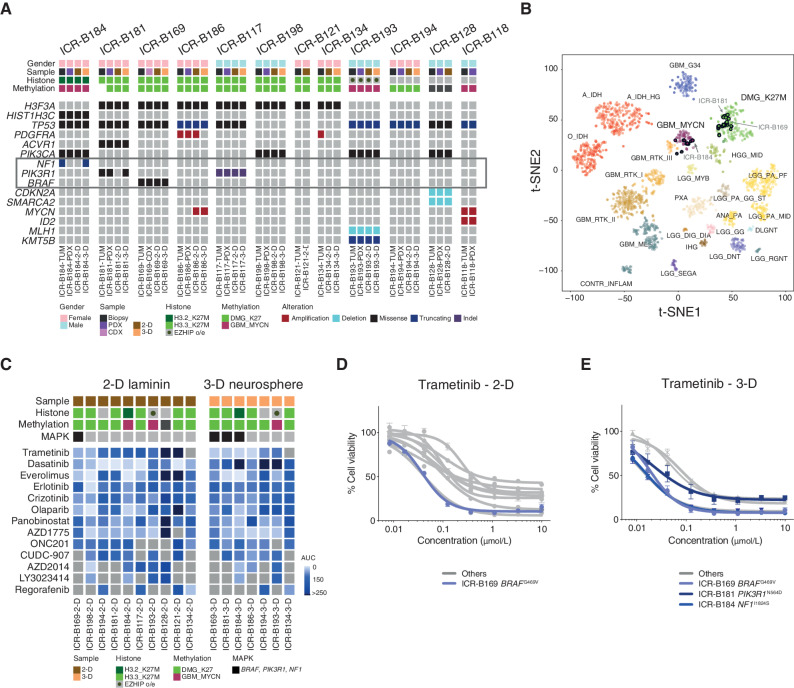
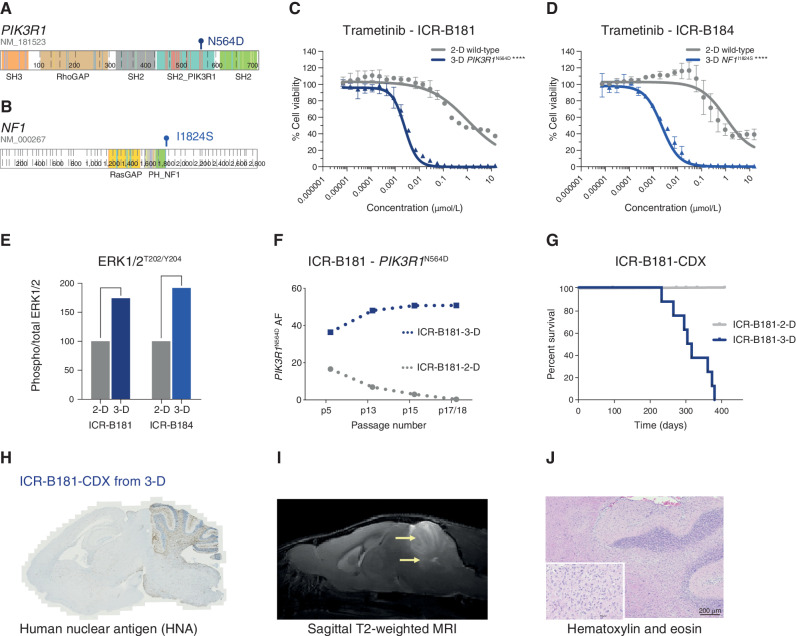
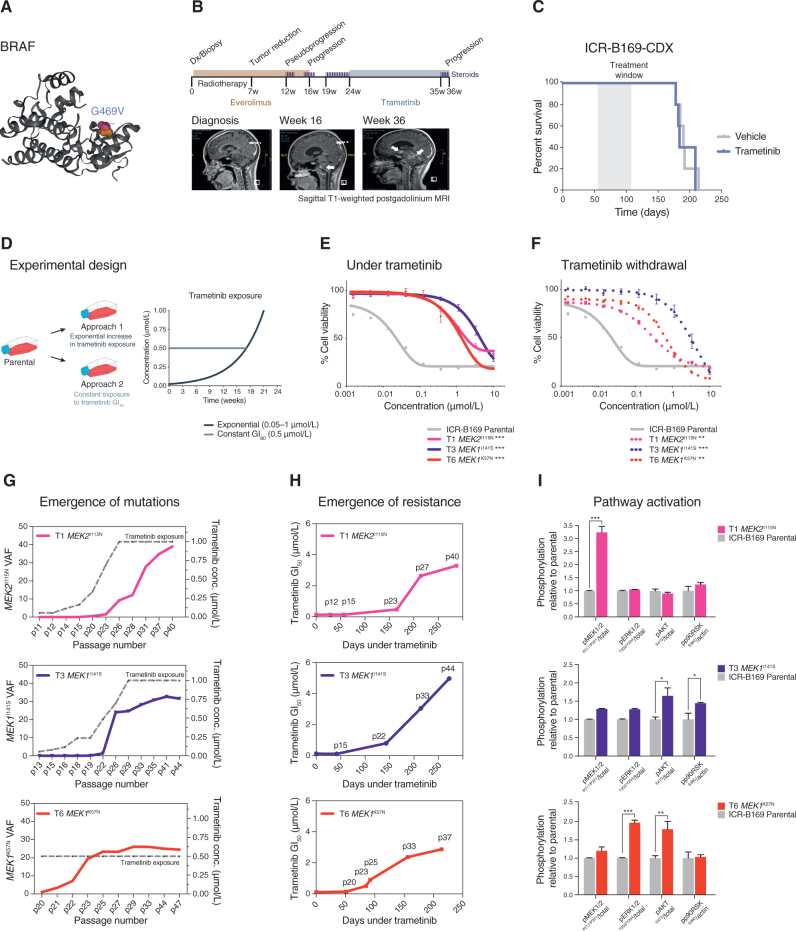
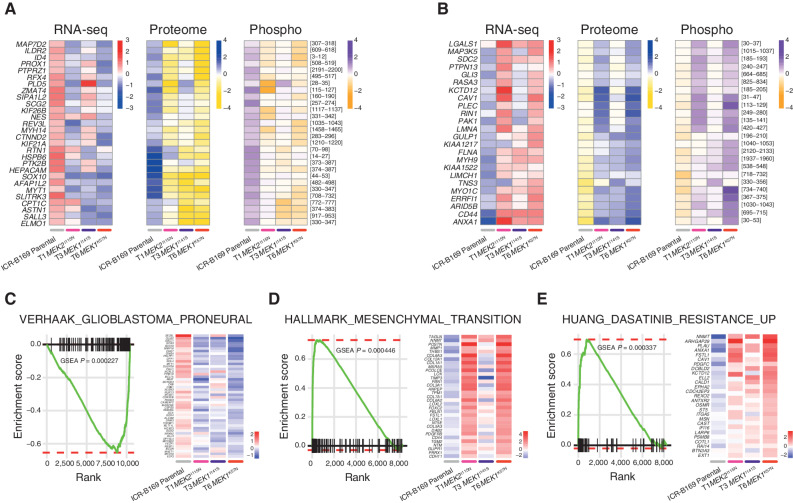
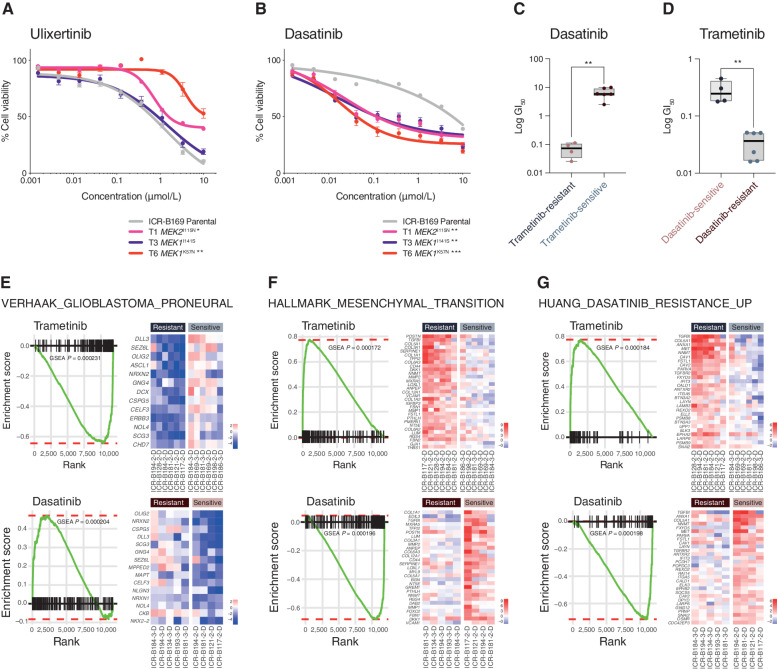
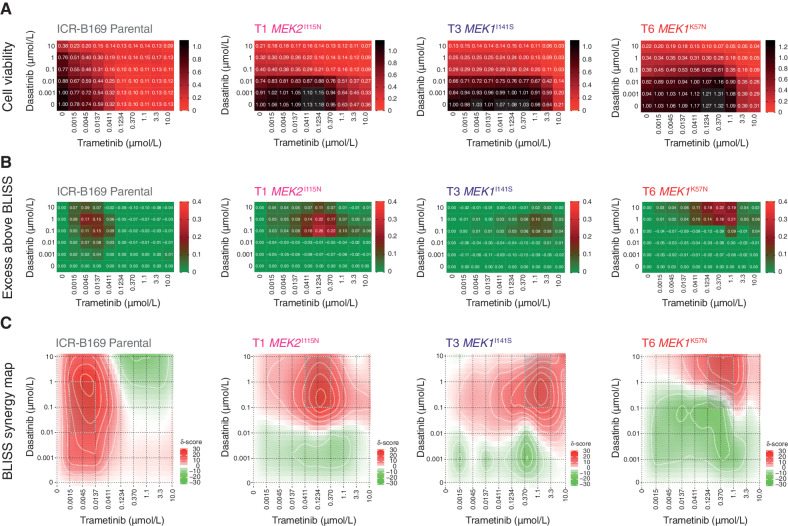
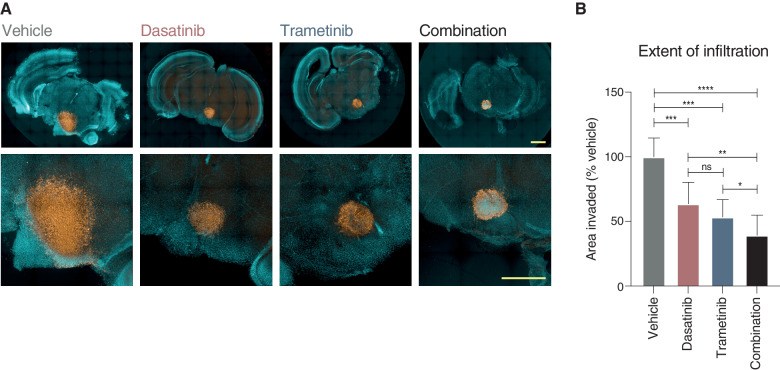
The survival of children with diffuse intrinsic pontine glioma (DIPG) remains dismal, with new treatments desperately needed. In a prospective biopsy-stratified clinical trial, we combined detailed molecular profiling and drug screening in newly established patient-derived models in vitro and in vivo. We identified in vitro sensitivity to MEK inhibitors in DIPGs harboring MAPK pathway alterations, but treatment of patient-derived xenograft models and a patient at relapse failed to elicit a significant response. We generated trametinib-resistant clones in a BRAFG469V model through continuous drug exposure and identified acquired mutations in MEK1/2 with sustained pathway upregulation. These cells showed hallmarks of mesenchymal transition and expression signatures overlapping with inherently trametinib-insensitive patient-derived cells, predicting sensitivity to dasatinib. Combined trametinib and dasatinib showed highly synergistic effects in vitro and on ex vivo brain slices. We highlight the MAPK pathway as a therapeutic target in DIPG and show the importance of parallel resistance modeling and combinatorial treatments for meaningful clinical translation.
Significance: We report alterations in the MAPK pathway in DIPGs to confer initial sensitivity to targeted MEK inhibition. We further identify for the first time the mechanism of resistance to single-agent targeted therapy in these tumors and suggest a novel combinatorial treatment strategy to overcome it in the clinic. This article is highlighted in the In This Issue feature, p. 587.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
- Cancer Discov. 12:587.
- Cancer Discov. 12:587.
References
-
- Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas Det al. . Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res 2001;56:127–55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
