

Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers
- PMID: 34737287
- PMCID: PMC8568941
- DOI: 10.1038/s41467-021-26600-5
Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers
Abstract
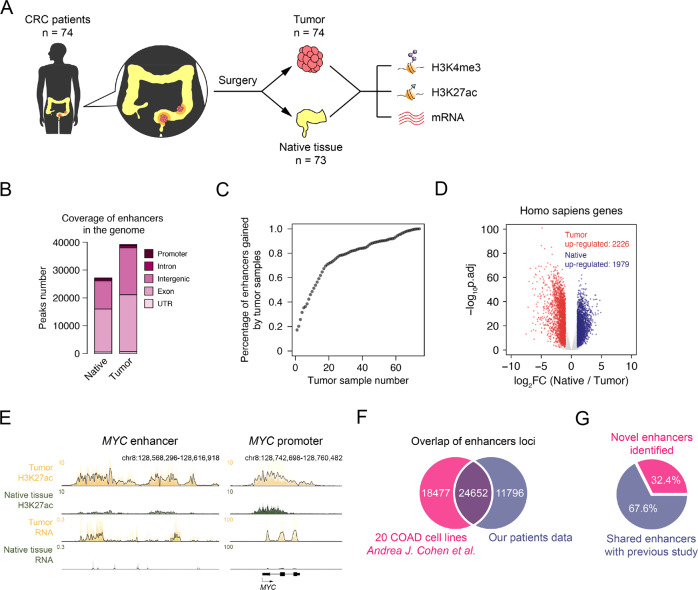
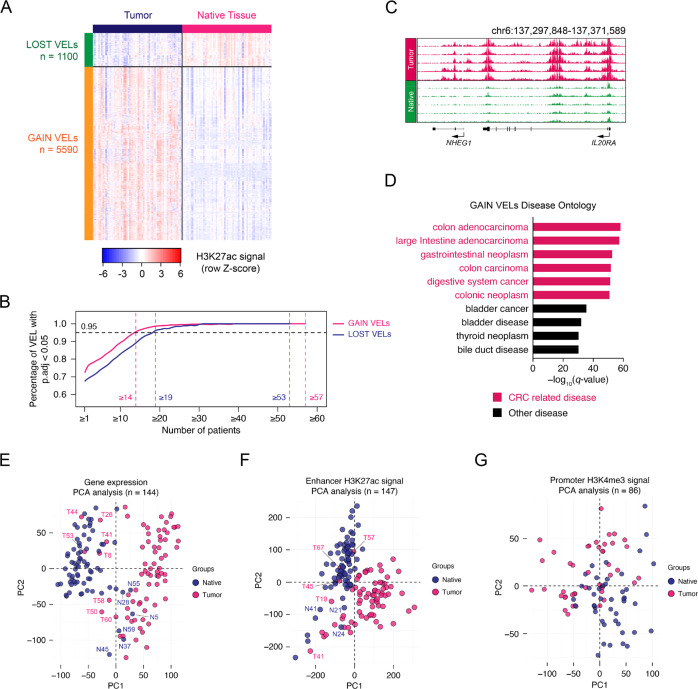
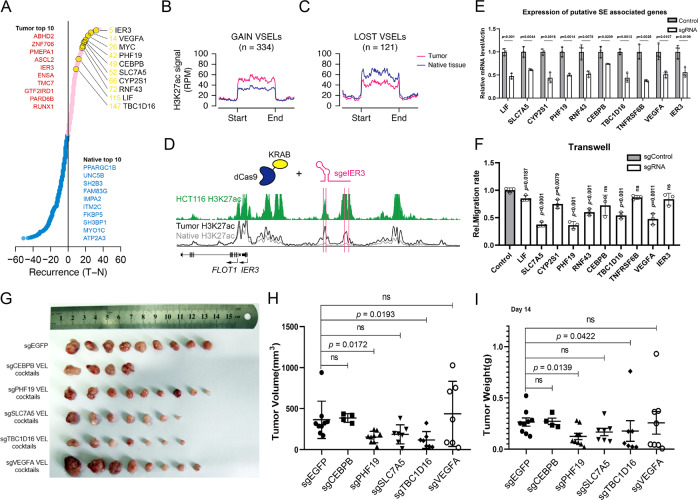
Colorectal cancer is one of the most common cancers in the world. Although genomic mutations and single nucleotide polymorphisms have been extensively studied, the epigenomic status in colorectal cancer patient tissues remains elusive. Here, together with genomic and transcriptomic analysis, we use ChIP-Seq to profile active enhancers at the genome wide level in colorectal cancer paired patient tissues (tumor and adjacent tissues from the same patients). In total, we sequence 73 pairs of colorectal cancer tissues and generate 147 H3K27ac ChIP-Seq, 144 RNA-Seq, 147 whole genome sequencing and 86 H3K4me3 ChIP-Seq samples. Our analysis identifies 5590 gain and 1100 lost variant enhancer loci in colorectal cancer, and 334 gain and 121 lost variant super enhancer loci. Multiple key transcription factors in colorectal cancer are predicted with motif analysis and core regulatory circuitry analysis. Further experiments verify the function of the super enhancers governing PHF19 and TBC1D16 in regulating colorectal cancer tumorigenesis, and KLF3 is identified as an oncogenic transcription factor in colorectal cancer. Taken together, our work provides an important epigenomic resource and functional factors for epigenetic studies in colorectal cancer.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
