

Elevating density functional theory to chemical accuracy for water simulations through a density-corrected many-body formalism
- PMID: 34737311
- PMCID: PMC8569147
- DOI: 10.1038/s41467-021-26618-9
Elevating density functional theory to chemical accuracy for water simulations through a density-corrected many-body formalism
Abstract
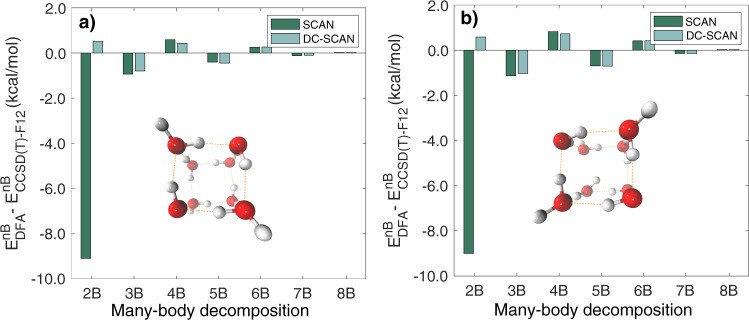
Density functional theory (DFT) has been extensively used to model the properties of water. Albeit maintaining a good balance between accuracy and efficiency, no density functional has so far achieved the degree of accuracy necessary to correctly predict the properties of water across the entire phase diagram. Here, we present density-corrected SCAN (DC-SCAN) calculations for water which, minimizing density-driven errors, elevate the accuracy of the SCAN functional to that of "gold standard" coupled-cluster theory. Building upon the accuracy of DC-SCAN within a many-body formalism, we introduce a data-driven many-body potential energy function, MB-SCAN(DC), that quantitatively reproduces coupled cluster reference values for interaction, binding, and individual many-body energies of water clusters. Importantly, molecular dynamics simulations carried out with MB-SCAN(DC) also reproduce the properties of liquid water, which thus demonstrates that MB-SCAN(DC) is effectively the first DFT-based model that correctly describes water from the gas to the liquid phase.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
-
- Franks, F. Water: A Matrix of Life Vol. 21 (Royal Society of Chemistry, 2000).
-
- Eisenberg, D., Kauzmann, W. & Kauzmann, W. The Structure and Properties of Water (Oxford University Press, 2005).
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
