

Design considerations for workflow management systems use in production genomics research and the clinic
- PMID: 34737383
- PMCID: PMC8569008
- DOI: 10.1038/s41598-021-99288-8
Design considerations for workflow management systems use in production genomics research and the clinic
Abstract
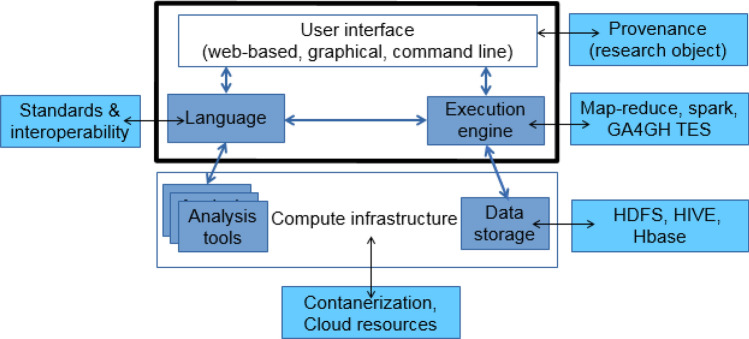
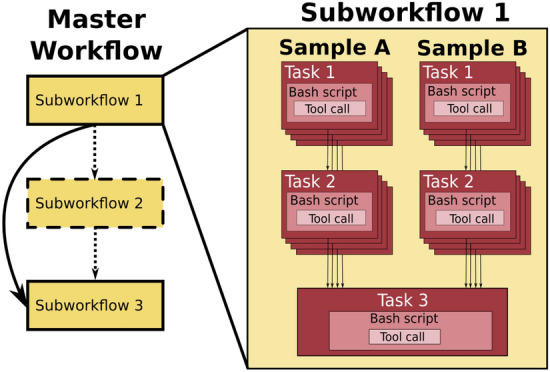
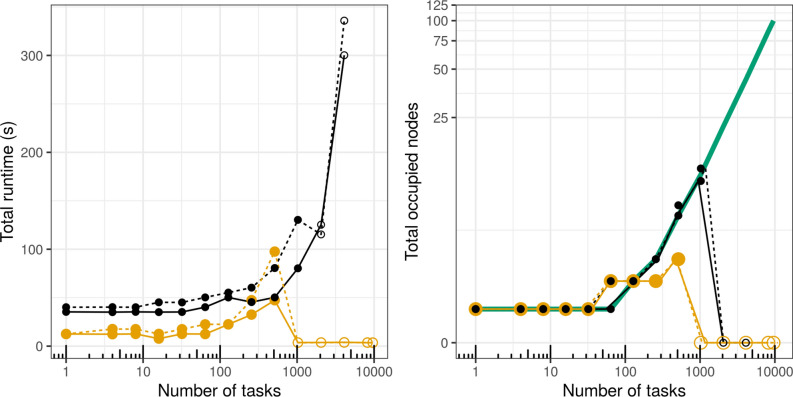
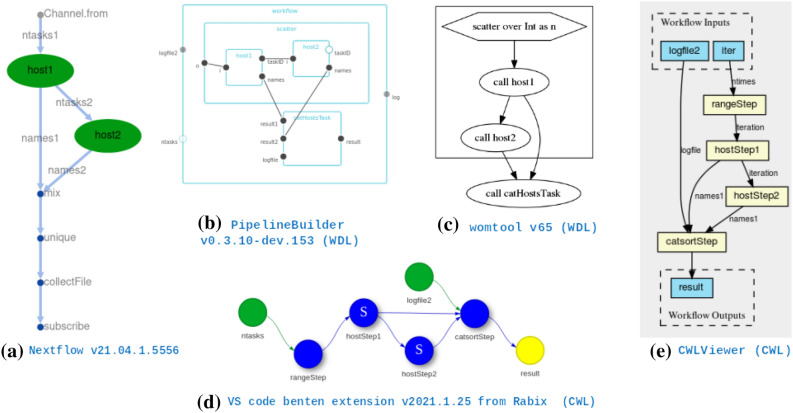
The changing landscape of genomics research and clinical practice has created a need for computational pipelines capable of efficiently orchestrating complex analysis stages while handling large volumes of data across heterogeneous computational environments. Workflow Management Systems (WfMSs) are the software components employed to fill this gap. This work provides an approach and systematic evaluation of key features of popular bioinformatics WfMSs in use today: Nextflow, CWL, and WDL and some of their executors, along with Swift/T, a workflow manager commonly used in high-scale physics applications. We employed two use cases: a variant-calling genomic pipeline and a scalability-testing framework, where both were run locally, on an HPC cluster, and in the cloud. This allowed for evaluation of those four WfMSs in terms of language expressiveness, modularity, scalability, robustness, reproducibility, interoperability, ease of development, along with adoption and usage in research labs and healthcare settings. This article is trying to answer, which WfMS should be chosen for a given bioinformatics application regardless of analysis type?. The choice of a given WfMS is a function of both its intrinsic language and engine features. Within bioinformatics, where analysts are a mix of dry and wet lab scientists, the choice is also governed by collaborations and adoption within large consortia and technical support provided by the WfMS team/community. As the community and its needs continue to evolve along with computational infrastructure, WfMSs will also evolve, especially those with permissive licenses that allow commercial use. In much the same way as the dataflow paradigm and containerization are now well understood to be very useful in bioinformatics applications, we will continue to see innovations of tools and utilities for other purposes, like big data technologies, interoperability, and provenance.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Deelman E, et al. The future of scientific workflows. Int. J. High Perform. Comput. Appl. 2017;32:159–175. doi: 10.1177/1094342017704893. - DOI
-
- Hines, J. Genomics code exceeds exaops on summit supercomputer: Oak ridge leadership computing facility (2018).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
