

RNA promotes the formation of spatial compartments in the nucleus
- PMID: 34739832
- PMCID: PMC9115877
- DOI: 10.1016/j.cell.2021.10.014
RNA promotes the formation of spatial compartments in the nucleus
Abstract
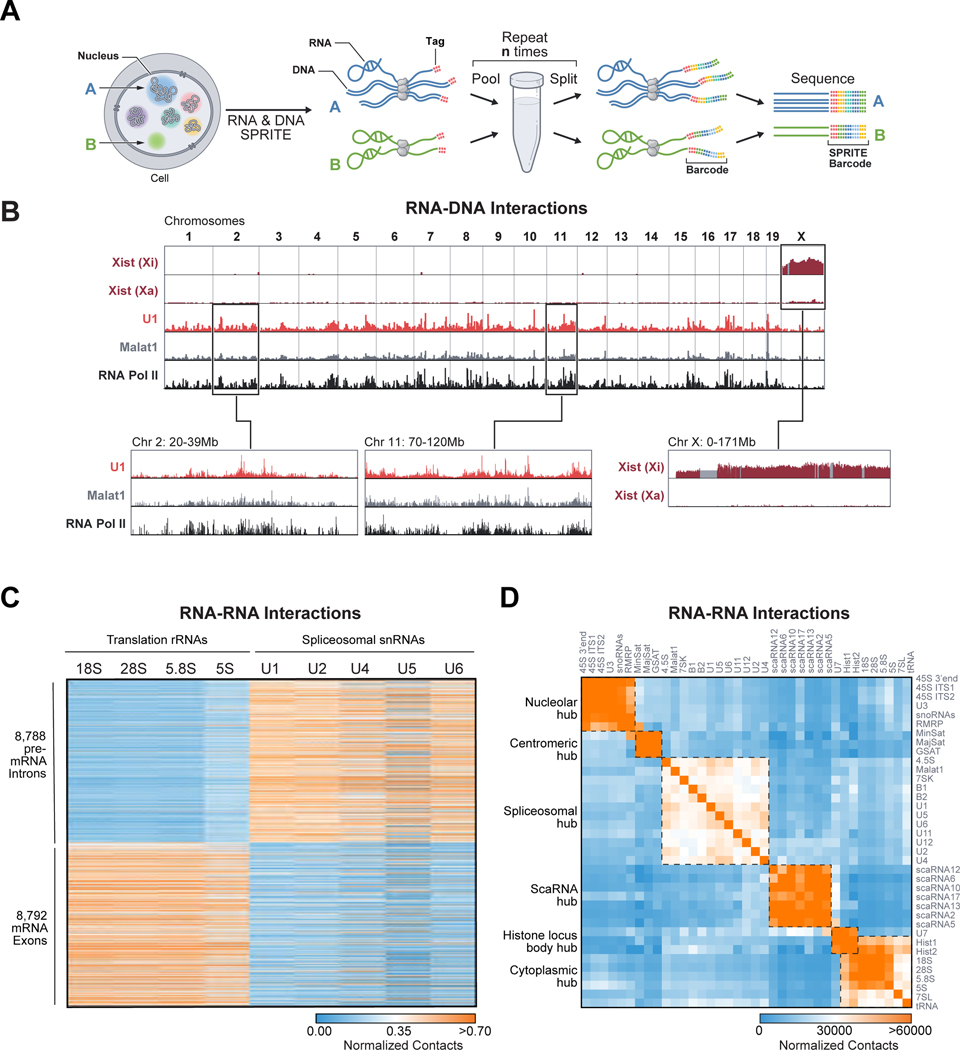
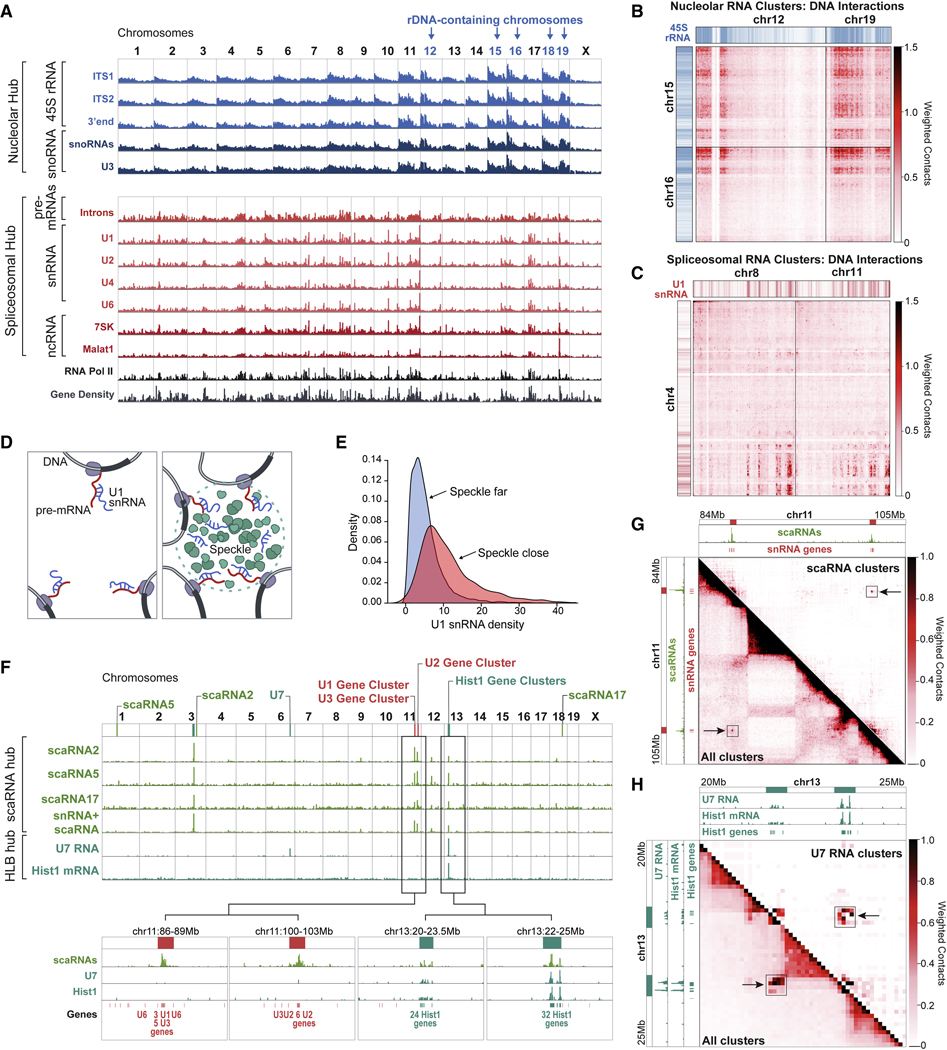
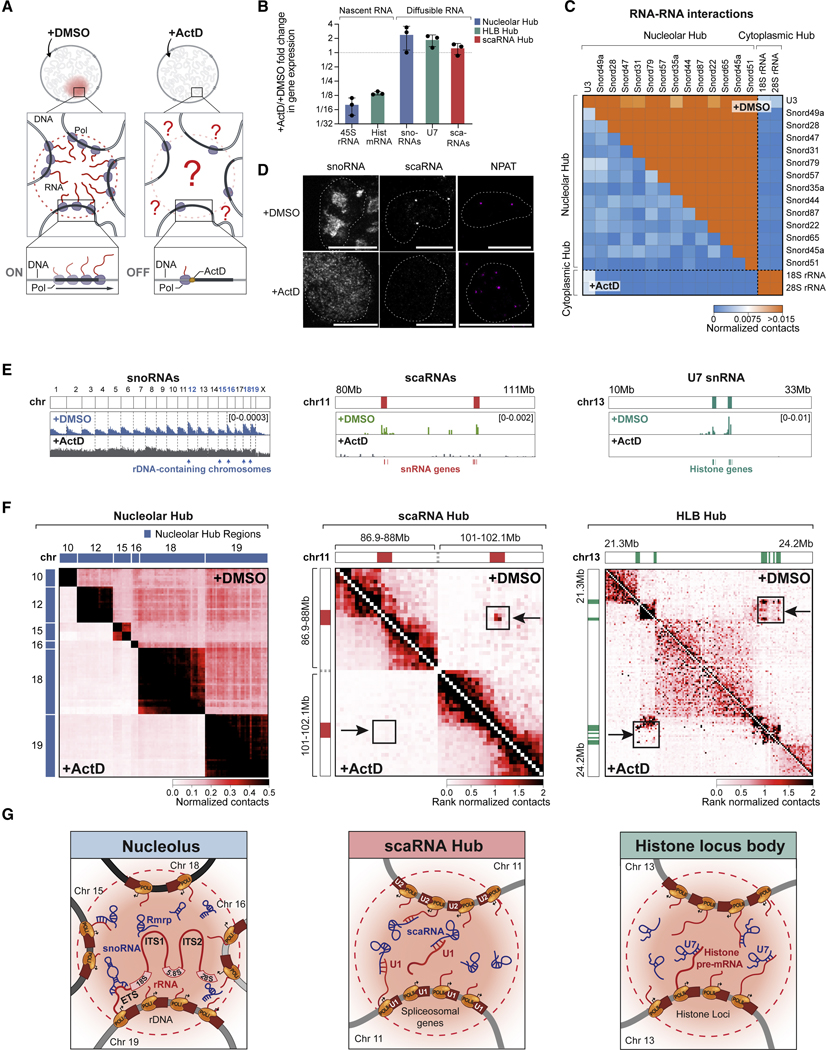
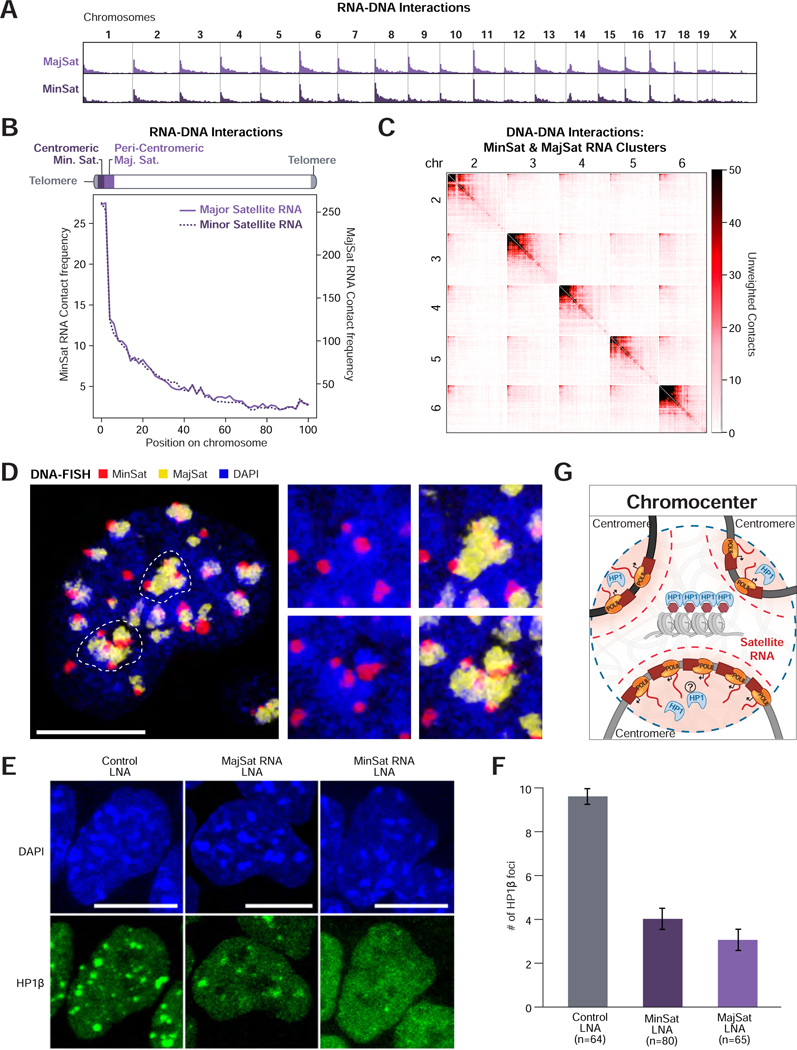
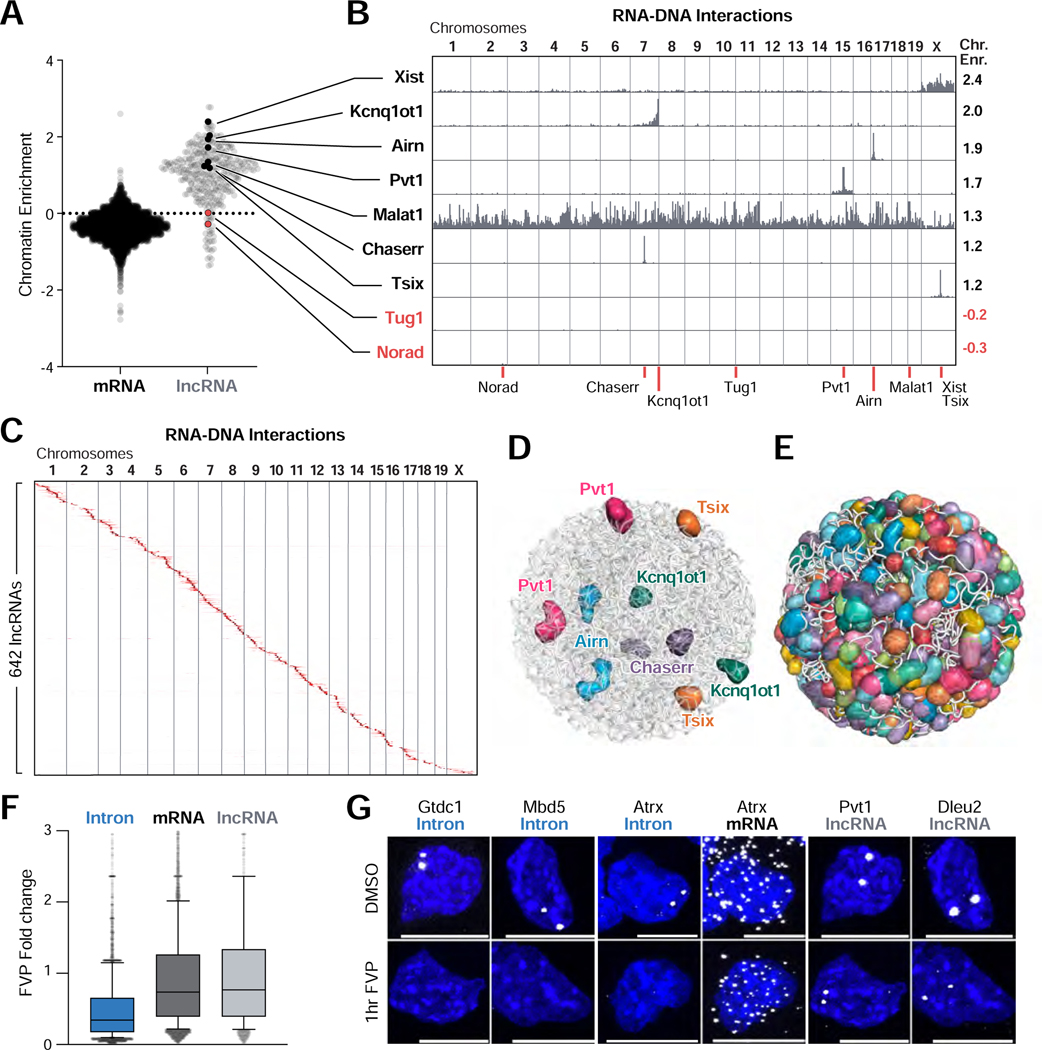
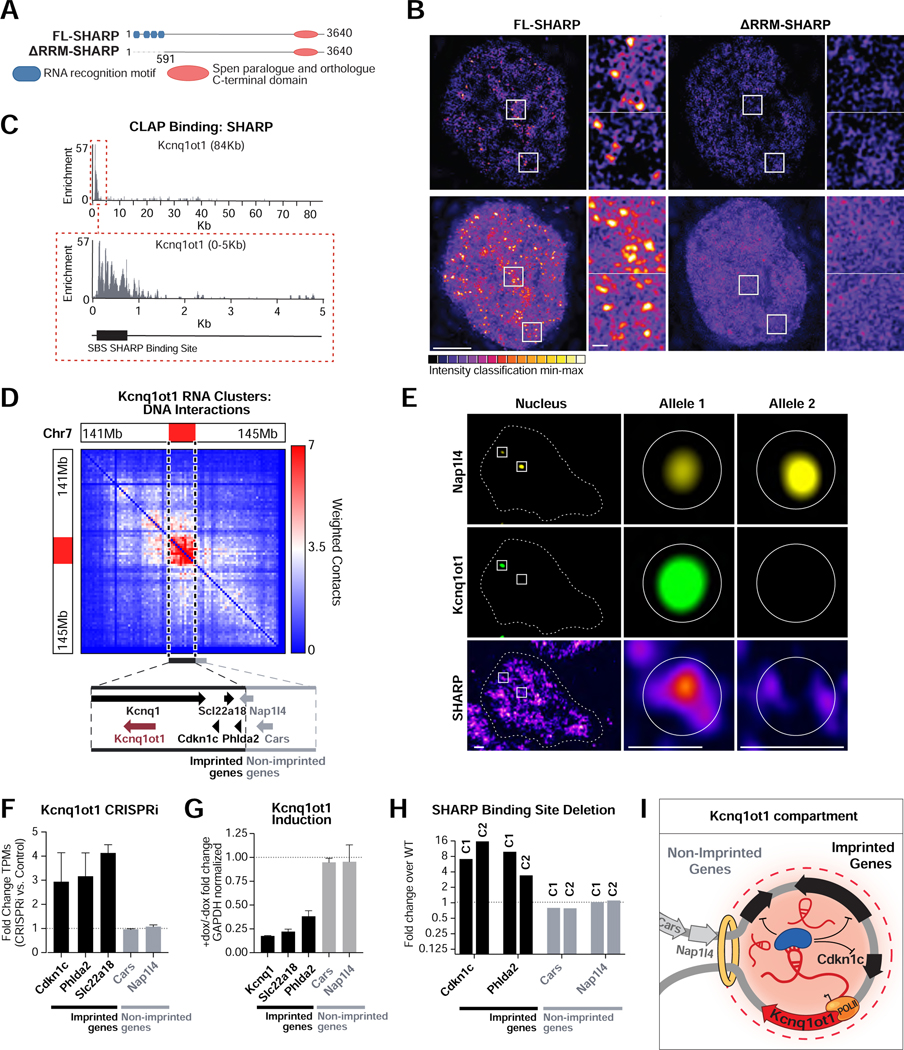
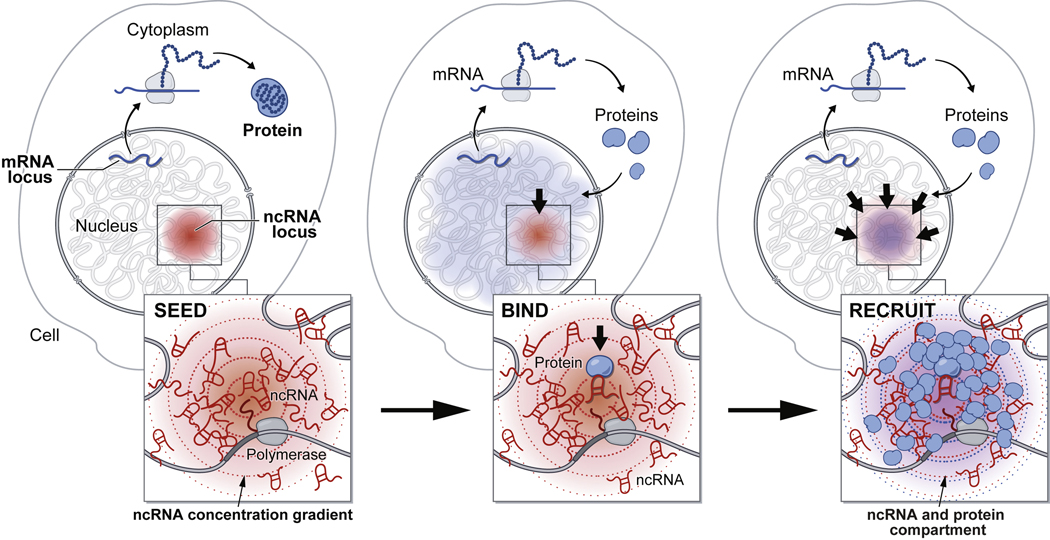
RNA, DNA, and protein molecules are highly organized within three-dimensional (3D) structures in the nucleus. Although RNA has been proposed to play a role in nuclear organization, exploring this has been challenging because existing methods cannot measure higher-order RNA and DNA contacts within 3D structures. To address this, we developed RNA & DNA SPRITE (RD-SPRITE) to comprehensively map the spatial organization of RNA and DNA. These maps reveal higher-order RNA-chromatin structures associated with three major classes of nuclear function: RNA processing, heterochromatin assembly, and gene regulation. These data demonstrate that hundreds of ncRNAs form high-concentration territories throughout the nucleus, that specific RNAs are required to recruit various regulators into these territories, and that these RNAs can shape long-range DNA contacts, heterochromatin assembly, and gene expression. These results demonstrate a mechanism where RNAs form high-concentration territories, bind to diffusible regulators, and guide them into compartments to regulate essential nuclear functions.
Keywords: RNA processing; cajal bodies; chromocenters; histone locus bodies; lncRNAs; ncRNAs; nuclear bodies; nuclear structure.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests S.A.Q. and M.G. are inventors on a patent covering the SPRITE method.
Figures
Comment in
-
Getting organized with non-coding RNAs.Nat Rev Genet. 2022 Jan;23(1):1. doi: 10.1038/s41576-021-00435-8. Nat Rev Genet. 2022. PMID: 34782779 No abstract available.
-
Looking for a job in a dynamic and collaborative working place? LncRNAs are recruiting!Cell. 2021 Dec 9;184(25):6019-6021. doi: 10.1016/j.cell.2021.10.020. Cell. 2021. PMID: 34890550
References
-
- Baßler J, and Hurt E. (2019). Eukaryotic Ribosome Assembly. Annu. Rev. Biochem - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
