

A multi-omic single-cell landscape of human gynecologic malignancies
- PMID: 34739872
- PMCID: PMC8642316
- DOI: 10.1016/j.molcel.2021.10.013
A multi-omic single-cell landscape of human gynecologic malignancies
Abstract
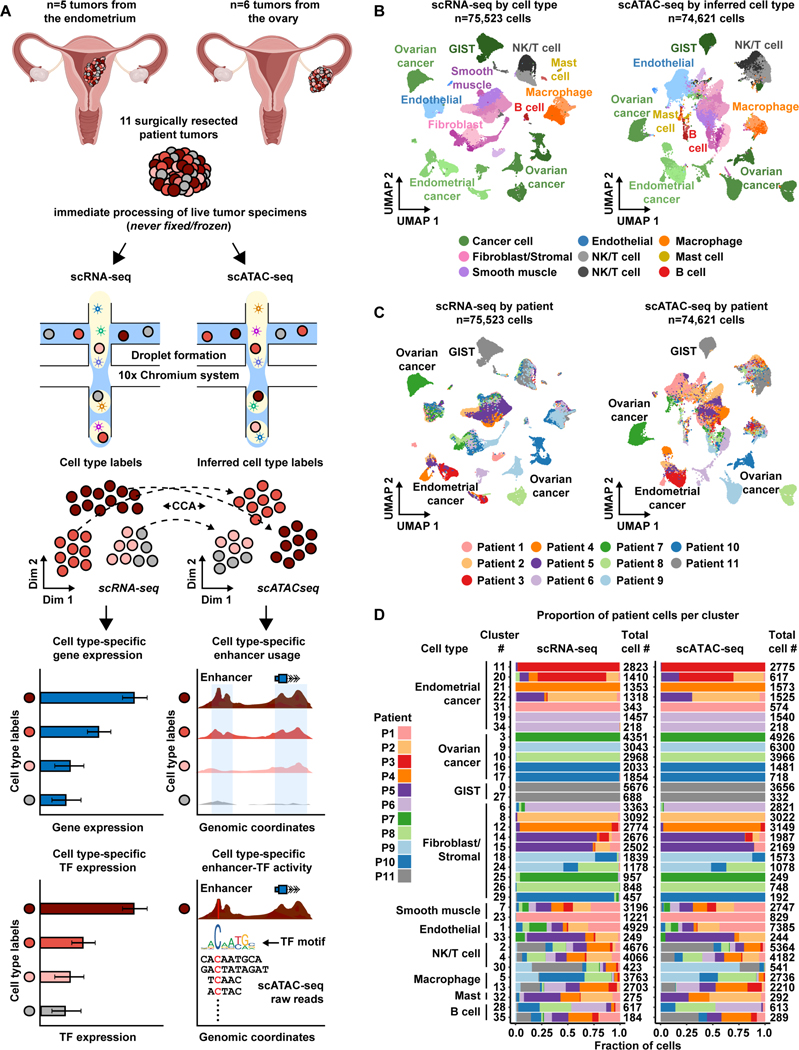
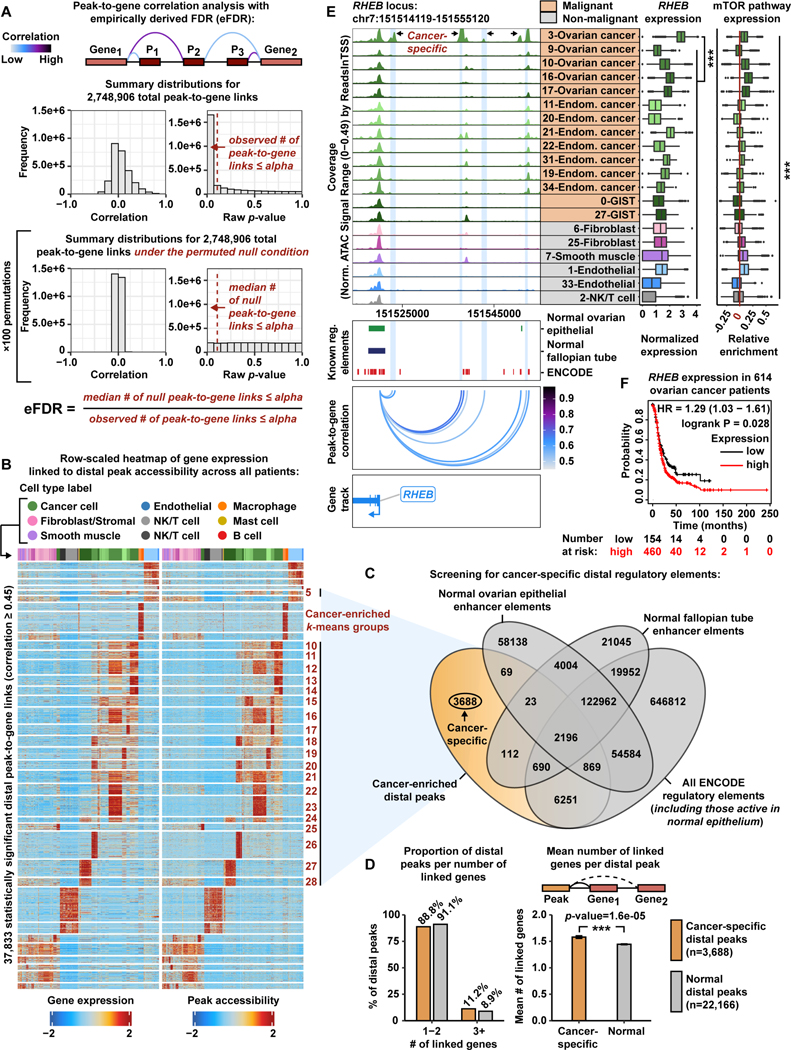
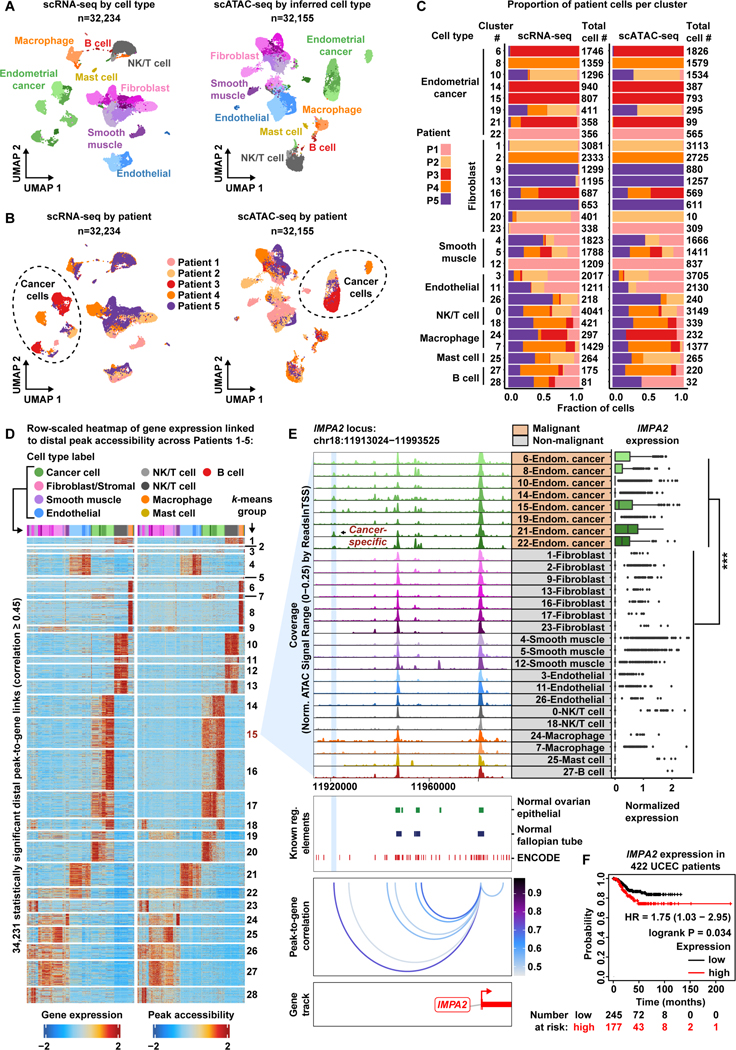
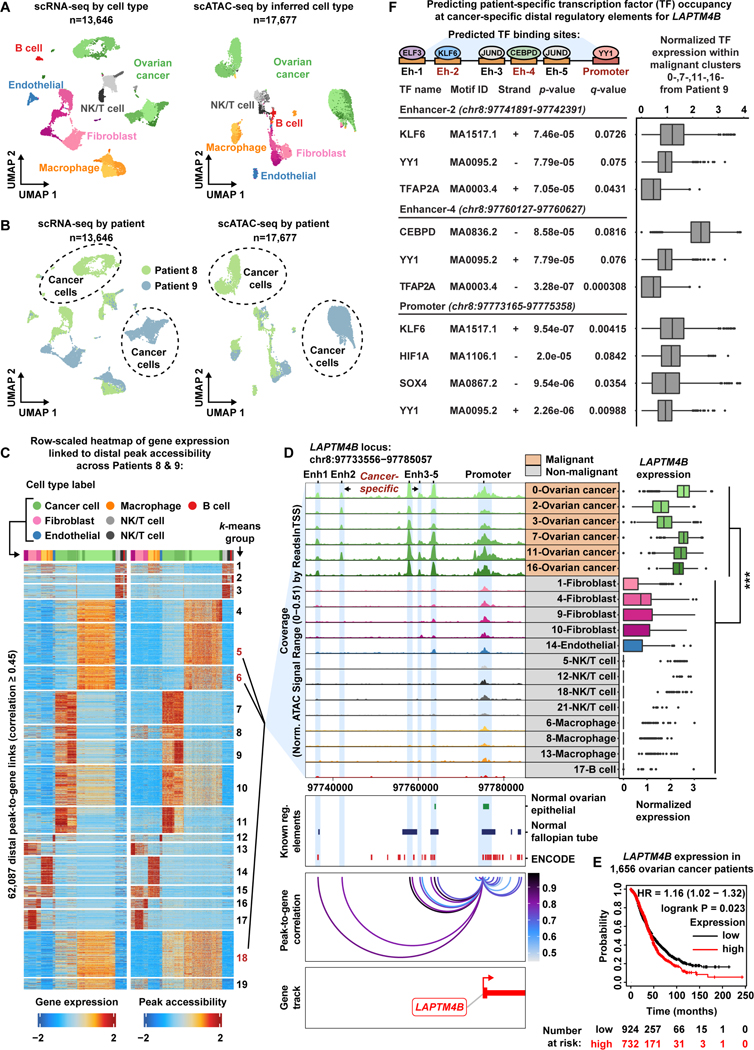
Deconvolution of regulatory mechanisms that drive transcriptional programs in cancer cells is key to understanding tumor biology. Herein, we present matched transcriptome (scRNA-seq) and chromatin accessibility (scATAC-seq) profiles at single-cell resolution from human ovarian and endometrial tumors processed immediately following surgical resection. This dataset reveals the complex cellular heterogeneity of these tumors and enabled us to quantitatively link variation in chromatin accessibility to gene expression. We show that malignant cells acquire previously unannotated regulatory elements to drive hallmark cancer pathways. Moreover, malignant cells from within the same patients show substantial variation in chromatin accessibility linked to transcriptional output, highlighting the importance of intratumoral heterogeneity. Finally, we infer the malignant cell type-specific activity of transcription factors. By defining the regulatory logic of cancer cells, this work reveals an important reliance on oncogenic regulatory elements and highlights the ability of matched scRNA-seq/scATAC-seq to uncover clinically relevant mechanisms of tumorigenesis in gynecologic cancers.
Keywords: chromatin accessibility; endometrial cancer; enhancer elements; gastro-intestinal stromal tumors; gene regulation; intratumoral heterogeneity; ovarian cancer; scATAC-seq; scRNA-seq; single-cell genomics.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
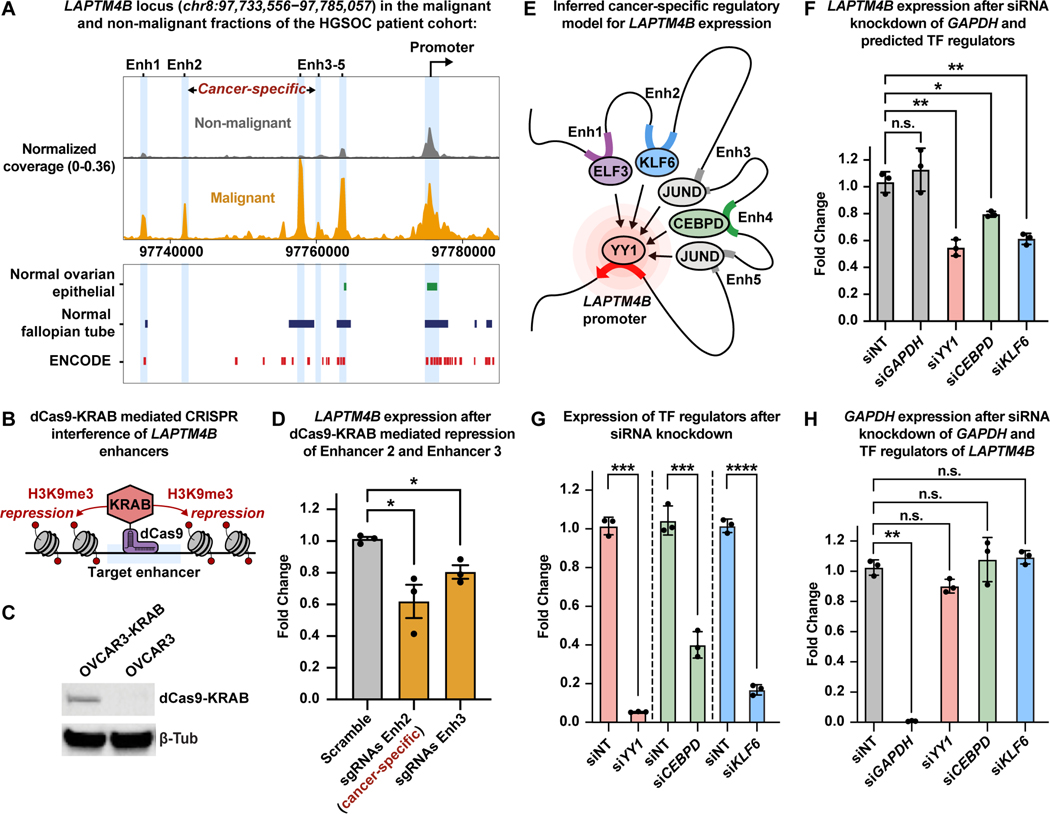
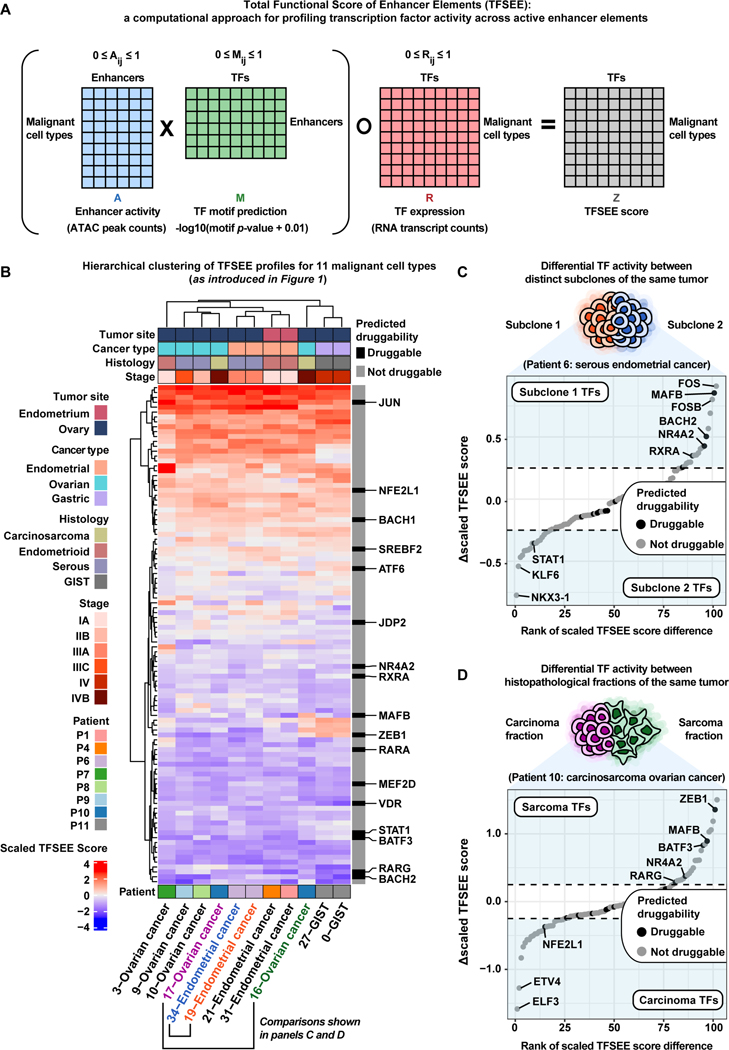
References
-
- BANERJI U 2014. A Phase I Trial of the Combination of AZD2014 and Weekly Paclitaxel [Online]. Available: https://ClinicalTrials.gov/show/NCT02193633 [Accessed].
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
