

De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations
- PMID: 34739968
- PMCID: PMC8545757
- DOI: 10.1016/j.compbiomed.2021.104967
De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations
Abstract
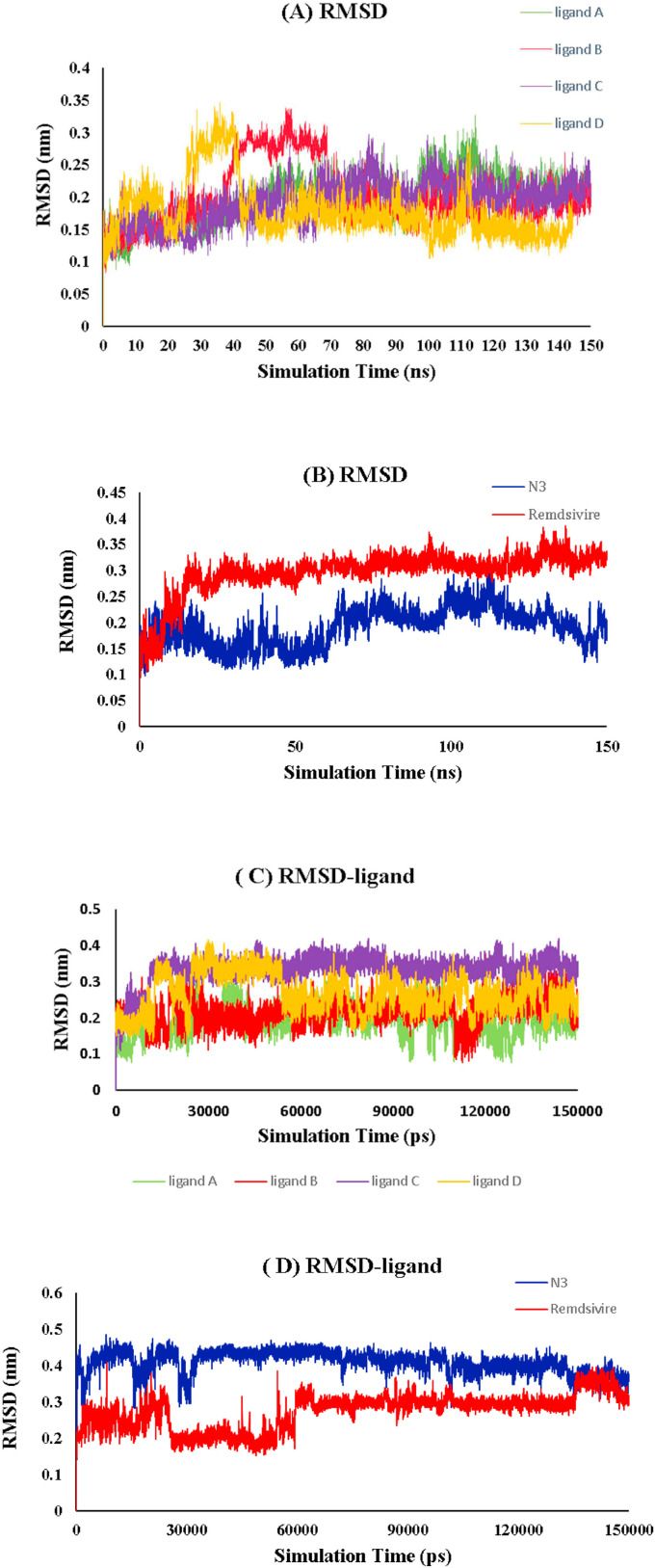
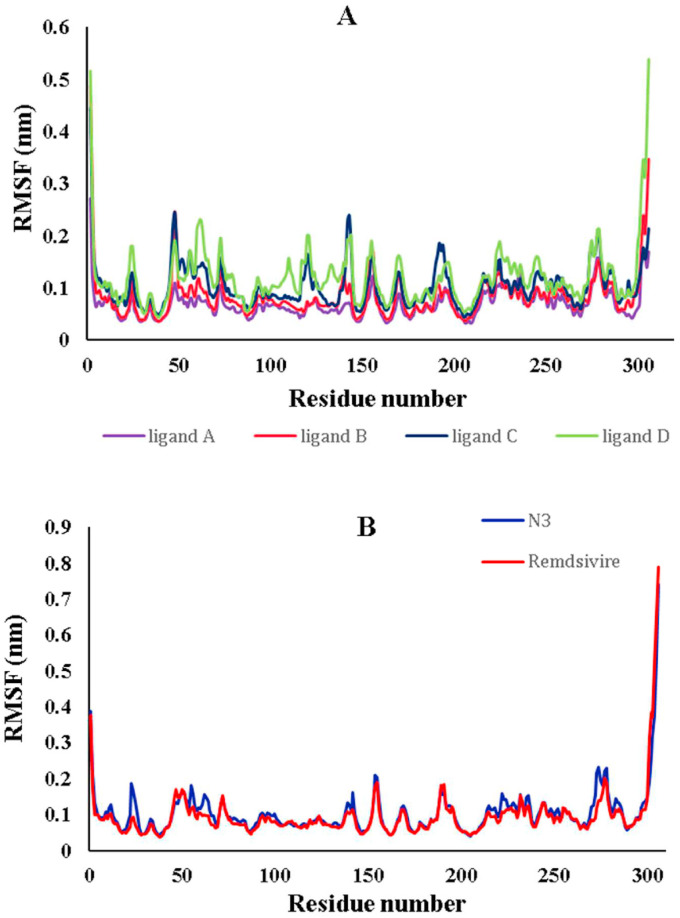
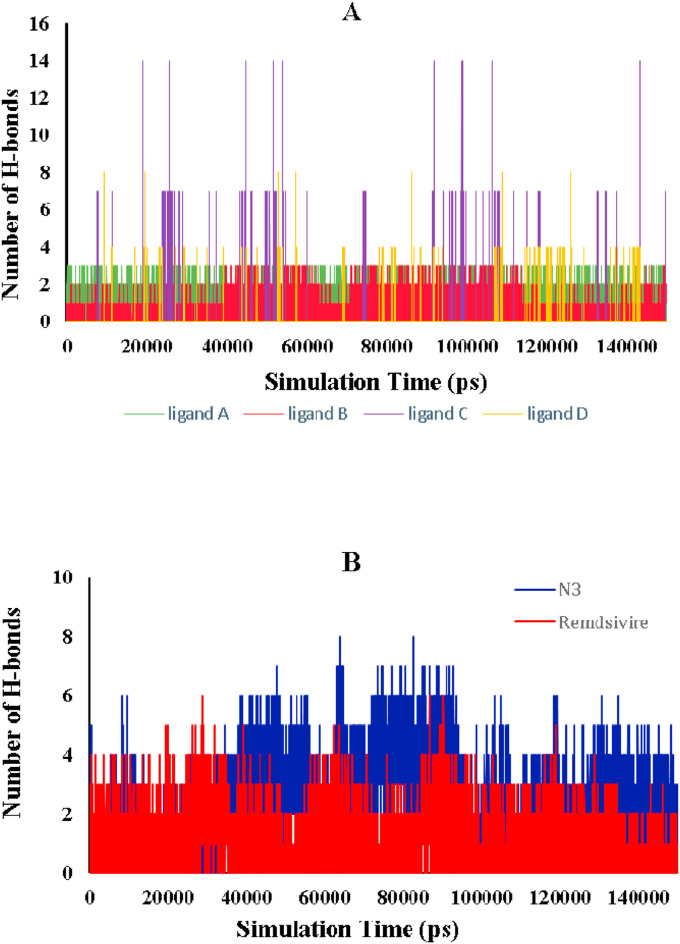
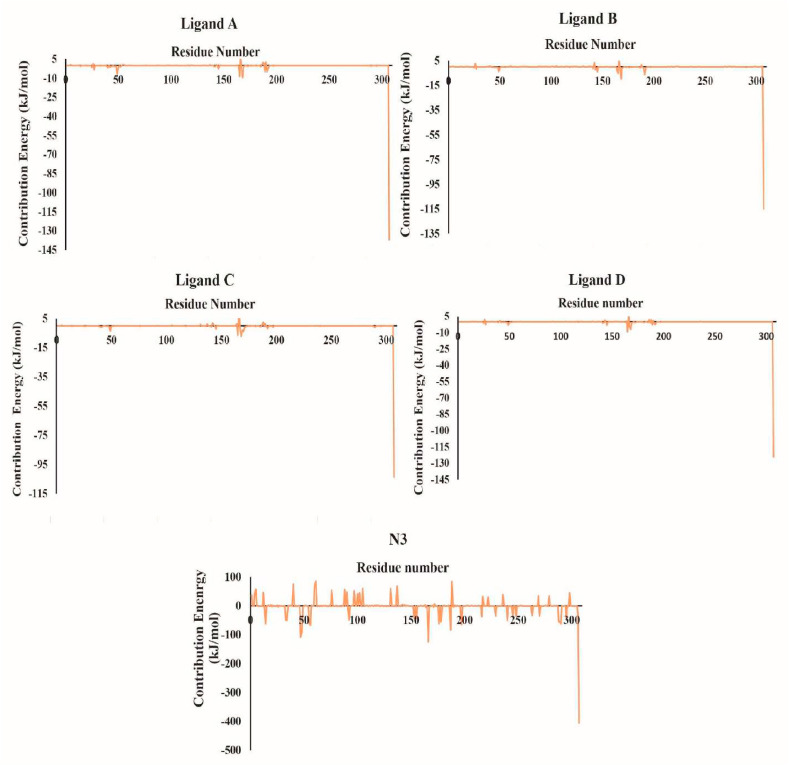
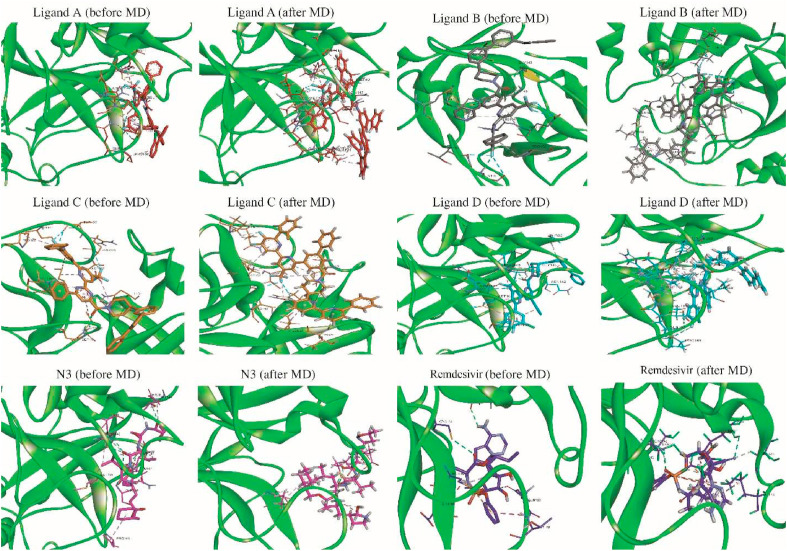
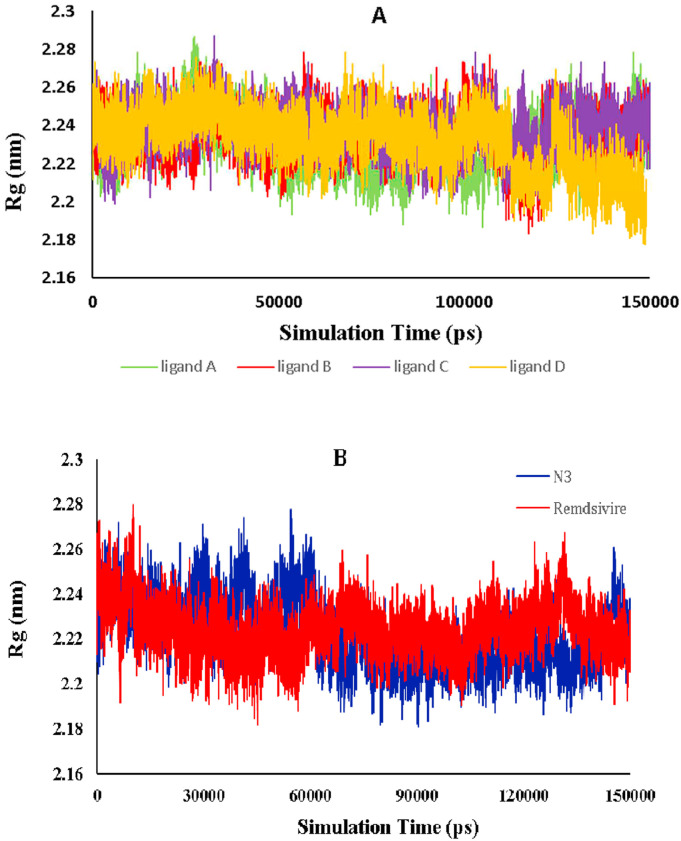
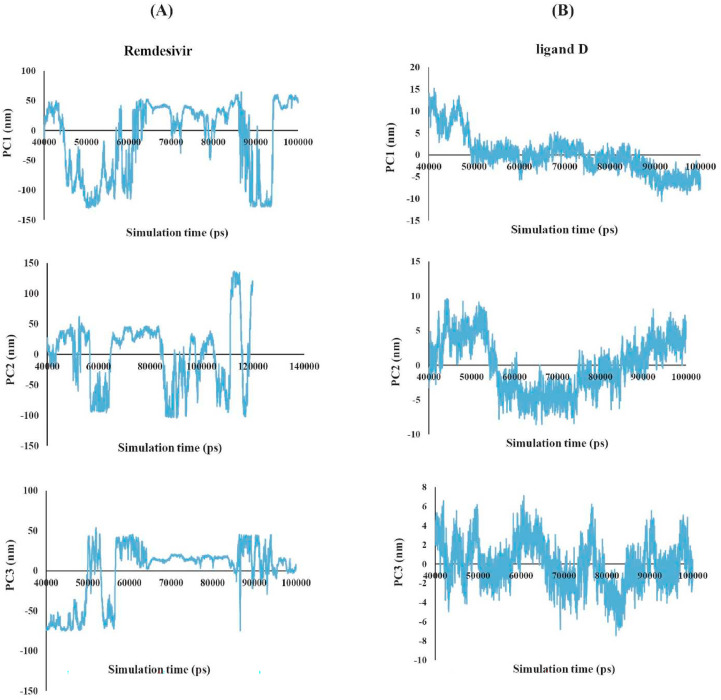
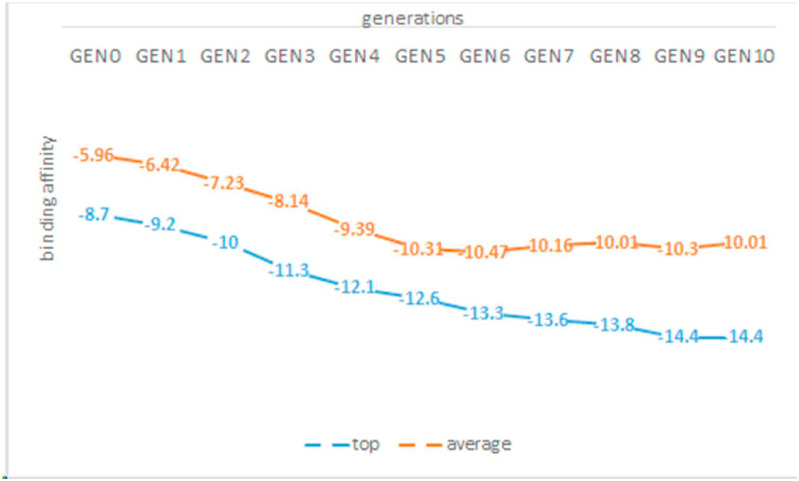
The main protease of SARS-CoV-2 is a critical target for the design and development of antiviral drugs. 2.5 M compounds were used in this study to train an LSTM generative network via transfer learning in order to identify the four best candidates capable of inhibiting the main proteases in SARS-CoV-2. The network was fine-tuned over ten generations, with each generation resulting in higher binding affinity scores. The binding affinities and interactions between the selected candidates and the SARS-CoV-2 main protease are predicted using a molecular docking simulation using AutoDock Vina. The compounds selected have a strong interaction with the key MET 165 and Cys145 residues. Molecular dynamics (MD) simulations were run for 150ns to validate the docking results on the top four ligands. Additionally, root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and hydrogen bond analysis strongly support these findings. Furthermore, the MM-PBSA free energy calculations revealed that these chemical molecules have stable and favorable energies, resulting in a strong binding with Mpro's binding site. This study's extensive computational and statistical analyses indicate that the selected candidates may be used as potential inhibitors against the SARS-CoV-2 in-silico environment. However, additional in-vitro, in-vivo, and clinical trials are required to demonstrate their true efficacy.
Keywords: Deep learning; Main protease; Molecular docking; Molecular dynamic simulation; SARS-CoV-2.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
None Declared.
Figures
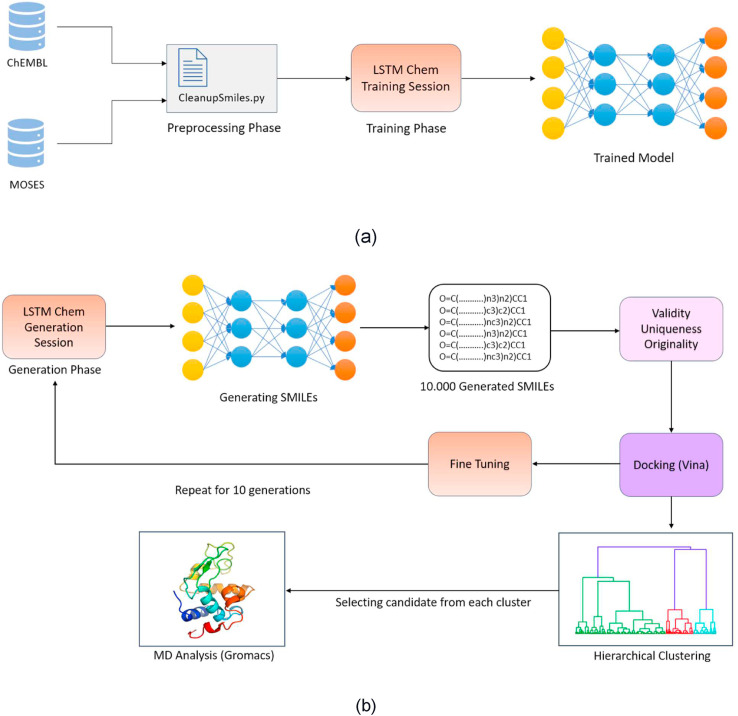
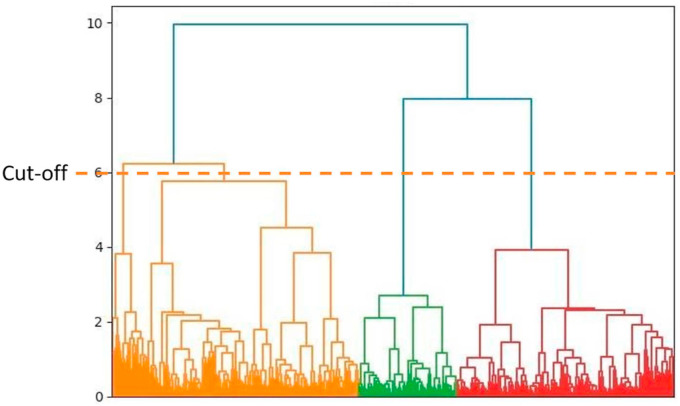
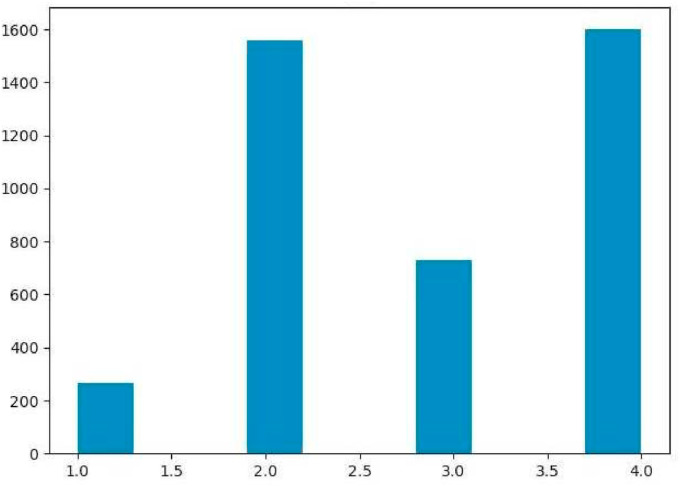
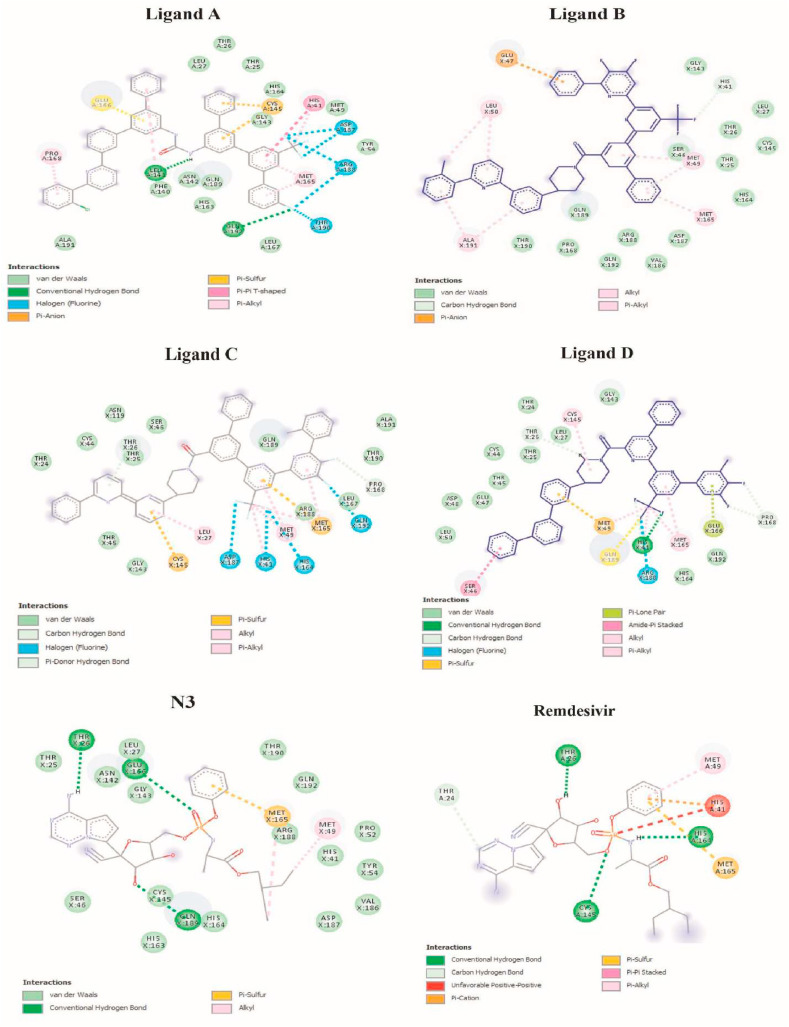
Similar articles
-
Some Flavolignans as Potent Sars-Cov-2 Inhibitors via Molecular Docking, Molecular Dynamic Simulations and ADME Analysis.Curr Comput Aided Drug Des. 2022;18(5):337-346. doi: 10.2174/1573409918666220816113516. Curr Comput Aided Drug Des. 2022. PMID: 35975852
-
A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2.J Biomol Struct Dyn. 2021 Jun;39(9):3213-3224. doi: 10.1080/07391102.2020.1761883. Epub 2020 May 12. J Biomol Struct Dyn. 2021. PMID: 32340562 Free PMC article.
-
Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site.Viruses. 2020 Aug 26;12(9):942. doi: 10.3390/v12090942. Viruses. 2020. PMID: 32859008 Free PMC article.
-
Characterizing the ligand-binding affinity toward SARS-CoV-2 Mpro via physics- and knowledge-based approaches.Phys Chem Chem Phys. 2022 Dec 14;24(48):29266-29278. doi: 10.1039/d2cp04476e. Phys Chem Chem Phys. 2022. PMID: 36449268 Review.
-
Molecular dynamics simulation approach for discovering potential inhibitors against SARS-CoV-2: A structural review.J Mol Liq. 2022 May 15;354:118901. doi: 10.1016/j.molliq.2022.118901. Epub 2022 Mar 9. J Mol Liq. 2022. PMID: 35309259 Free PMC article. Review.
Cited by
-
6-Bromo quinazoline derivatives as cytotoxic agents: design, synthesis, molecular docking and MD simulation.BMC Chem. 2024 Jul 4;18(1):125. doi: 10.1186/s13065-024-01230-2. BMC Chem. 2024. PMID: 38965630 Free PMC article.
-
Investigation of the binding and dynamic features of A.30 variant revealed higher binding of RBD for hACE2 and escapes the neutralizing antibody: A molecular simulation approach.Comput Biol Med. 2022 Jul;146:105574. doi: 10.1016/j.compbiomed.2022.105574. Epub 2022 Apr 30. Comput Biol Med. 2022. PMID: 35533461 Free PMC article.
-
E2UbcH5B-derived peptide ligands target HECT E3-E2 binding site and block the Ub-dependent SARS-CoV-2 egression: A computational study.Comput Biol Med. 2022 Jul;146:105660. doi: 10.1016/j.compbiomed.2022.105660. Epub 2022 May 22. Comput Biol Med. 2022. PMID: 35751189 Free PMC article.
-
Identification of SARS-CoV-2 Mpro inhibitors through deep reinforcement learning for de novo drug design and computational chemistry approaches.RSC Med Chem. 2024 Apr 29;15(6):2146-2159. doi: 10.1039/d4md00106k. eCollection 2024 Jun 19. RSC Med Chem. 2024. PMID: 38911172 Free PMC article.
-
Multi-omics analysis of the anti-cancer effects of curcumol in endometrial carcinoma.Front Pharmacol. 2025 Jul 3;16:1565959. doi: 10.3389/fphar.2025.1565959. eCollection 2025. Front Pharmacol. 2025. PMID: 40678730 Free PMC article.
References
-
- Prevention Centers for Disease Control and. Novel Coronavirus. Information for Healthcare Professionals; Wuhan China: 2019.
-
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003 Jun 13;300(5626):1763–1767. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
