

Advances in integrative African genomics
- PMID: 34740451
- PMCID: PMC8752515
- DOI: 10.1016/j.tig.2021.09.013
Advances in integrative African genomics
Abstract
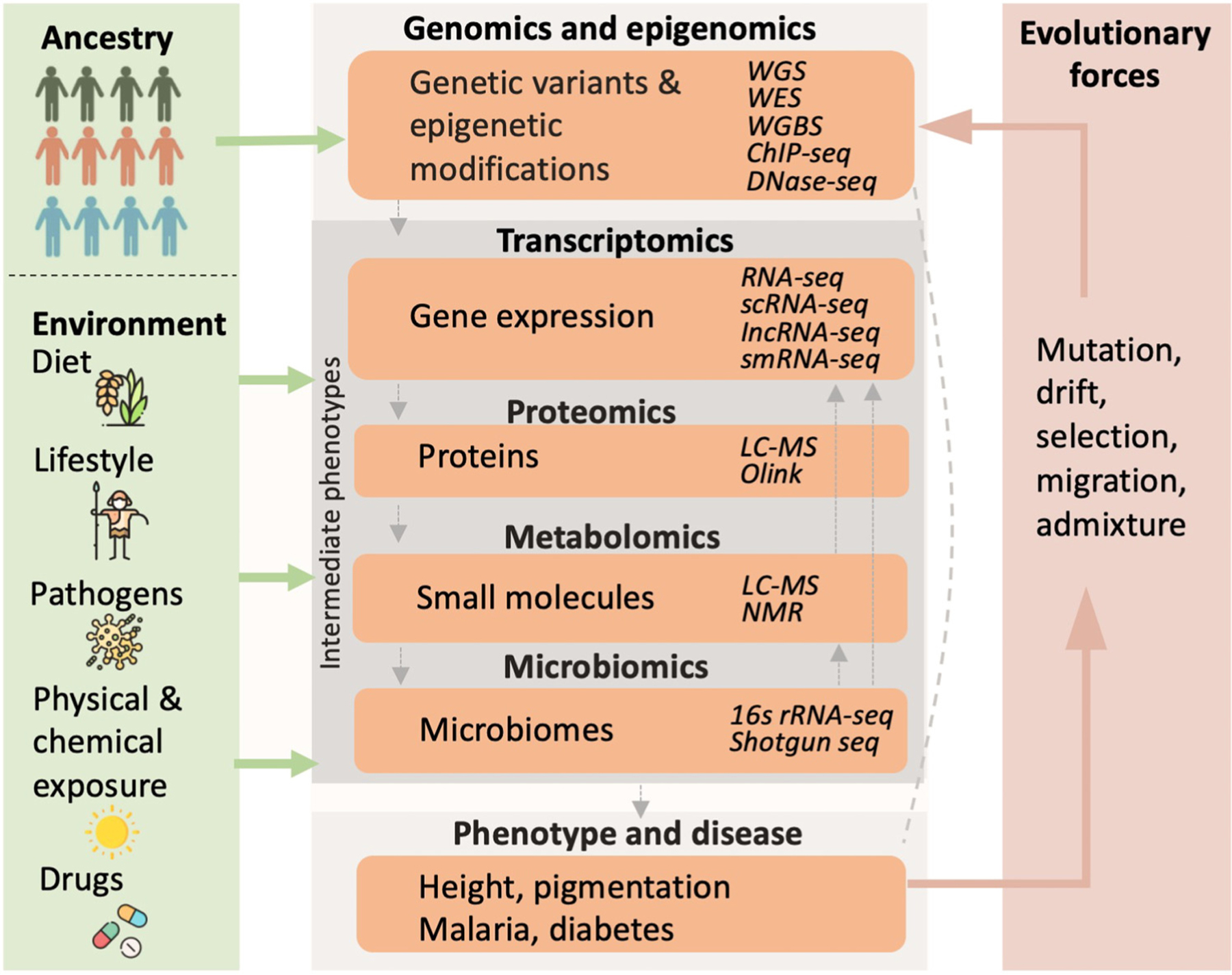
There has been a rapid increase in human genome sequencing in the past two decades, resulting in the identification of millions of previously unknown genetic variants. However, African populations are under-represented in sequencing efforts. Additional sequencing from diverse African populations and the construction of African-specific reference genomes is needed to better characterize the full spectrum of variation in humans. However, sequencing alone is insufficient to address the molecular and cellular mechanisms underlying variable phenotypes and disease risks. Determining functional consequences of genetic variation using multi-omics approaches is a fundamental post-genomic challenge. We discuss approaches to close the knowledge gaps about African genomic diversity and review advances in African integrative genomic studies and their implications for precision medicine.
Keywords: Africans; genomic diversity; integrative genomics; intermediate phenotype; omics; population-specific reference genome.
Copyright © 2021. Published by Elsevier Ltd.
Conflict of interest statement
Declaration of interests
No interests are declared.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
