

Mucopolysaccharidosis III: Molecular basis and treatment
- PMID: 34743503
- PMCID: PMC10228206
- DOI: 10.5114/pedm.2021.109270
Mucopolysaccharidosis III: Molecular basis and treatment
Abstract
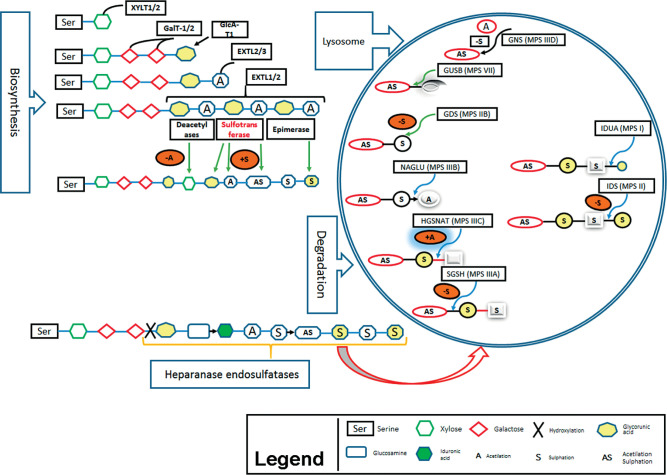
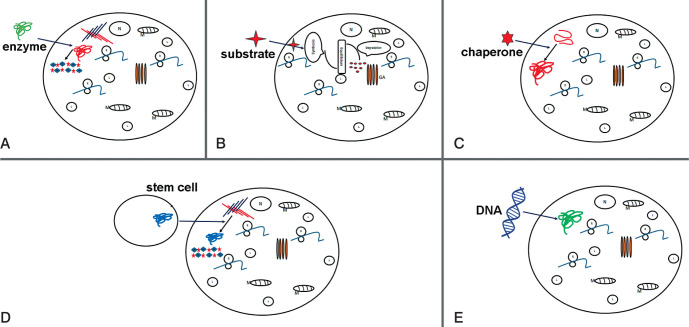
Mucopolysaccharidoses (MPSs) are known as rare genetic diseases which are caused by mutation in the enzyme heparin sulfate, which normally leads to degradation and accumulation of glycosaminoglycans in the cells. There are 11 types of MPSs, whereby neuropathy may occur in seven of them (MPS I, II, IIIA, IIIB, IIIC, IIID and VII). Accumulation of degraded heparin sulfate in lysosomes causes cellular dysfunction and malfunction of several organs. However, the exact molecular mechanism how protein degradation and storage leads to cellular dysfunction is not understood, yet. Nonetheless, several genetic and biochemical methods for diagnosis of MPSs are available nowadays. Here we provide an overview on known molecular basis of MPS in general, including enzyme defects and symptoms of MPS; however, the main focus is on MPS type III together with potential and perspective therapy-options.
Keywords: clinical diagnosis.; glycosaminoglycan; lysosomal storage diseases (LSDs); therapy for mucopolysccharidosis; mucopolysaccharidosis III (MPS III).
Conflict of interest statement
none declared.
Figures
Similar articles
-
Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III.Mol Genet Metab. 2012 Dec;107(4):705-10. doi: 10.1016/j.ymgme.2012.09.024. Epub 2012 Sep 28. Mol Genet Metab. 2012. PMID: 23084433
-
Glycosaminoglycans and mucopolysaccharidosis type III.Front Biosci (Landmark Ed). 2016 Jun 1;21(7):1393-409. doi: 10.2741/4463. Front Biosci (Landmark Ed). 2016. PMID: 27100513 Review.
-
Validation of Liquid Chromatography-Tandem Mass Spectrometry-Based 5-Plex Assay for Mucopolysaccharidoses.Int J Mol Sci. 2020 Mar 16;21(6):2025. doi: 10.3390/ijms21062025. Int J Mol Sci. 2020. PMID: 32188102 Free PMC article.
-
Pathogenic Roles of Heparan Sulfate and Its Use as a Biomarker in Mucopolysaccharidoses.Int J Mol Sci. 2022 Oct 3;23(19):11724. doi: 10.3390/ijms231911724. Int J Mol Sci. 2022. PMID: 36233030 Free PMC article. Review.
-
Evaluation of heparin cofactor II-thrombin complex as a biomarker on blood spots from mucopolysaccharidosis I, IIIA and IIIB mice.Mol Genet Metab. 2010 Mar;99(3):269-74. doi: 10.1016/j.ymgme.2009.10.175. Epub 2009 Oct 23. Mol Genet Metab. 2010. PMID: 19926322
Cited by
-
Rare Presentation of Attenuated Mucopolysaccharidosis Type IIIA as Isolated Retinitis Pigmentosa.J Vitreoretin Dis. 2025 May 10:24741264251340108. doi: 10.1177/24741264251340108. Online ahead of print. J Vitreoretin Dis. 2025. PMID: 40357345 Free PMC article.
-
Highly diverse phenotypes of mucopolysaccharidosis type IIIB sibling patients: effects of an additional mutation in the AUTS2 gene.J Appl Genet. 2022 Sep;63(3):535-542. doi: 10.1007/s13353-022-00702-2. Epub 2022 May 8. J Appl Genet. 2022. PMID: 35525889
-
Structure and mechanism of lysosome transmembrane acetylation by HGSNAT.Nat Struct Mol Biol. 2024 Oct;31(10):1502-1508. doi: 10.1038/s41594-024-01315-5. Epub 2024 May 20. Nat Struct Mol Biol. 2024. PMID: 38769387
-
Various AAV Serotypes and Their Applications in Gene Therapy: An Overview.Cells. 2023 Mar 1;12(5):785. doi: 10.3390/cells12050785. Cells. 2023. PMID: 36899921 Free PMC article. Review.
-
Identification and characterization of novel genetic variants in the first Chinese family of mucopolysaccharidosis IIIC (Sanfilippo C syndrome).J Cell Mol Med. 2024 Apr;28(8):e18307. doi: 10.1111/jcmm.18307. J Cell Mol Med. 2024. PMID: 38613342 Free PMC article.
References
-
- Tomatsu S, Lavery C, Giugliani R, et al. . Mucopolysaccharidoses Update; Nova Science Publishers: Hauppauge, NY, USA: 2018.
-
- Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Valle DL, Antonarakis S, et al.. (eds.). McGraw-Hill, New York, NY, USA, 2001; 3421–3452.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources