

Contamination in Reference Sequence Databases: Time for Divide-and-Rule Tactics
- PMID: 34745061
- PMCID: PMC8570097
- DOI: 10.3389/fmicb.2021.755101
Contamination in Reference Sequence Databases: Time for Divide-and-Rule Tactics
Abstract
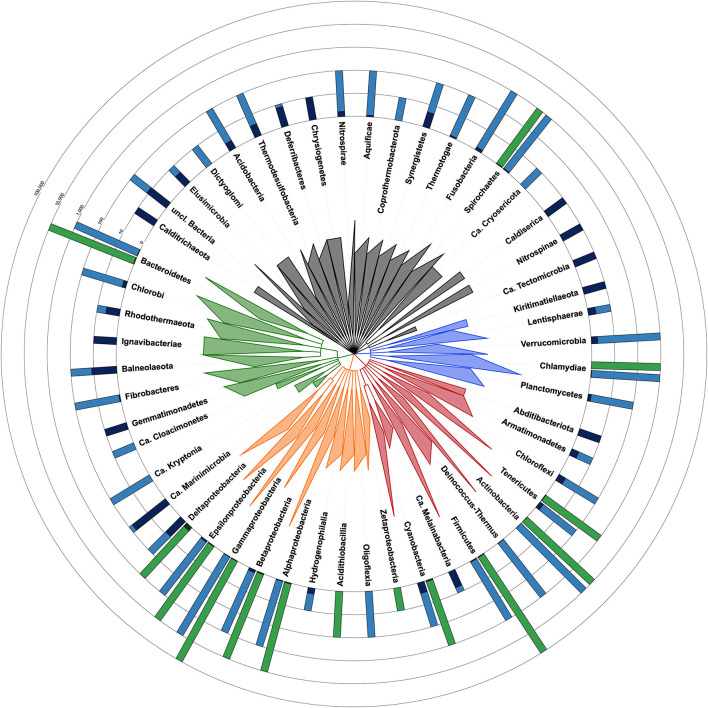
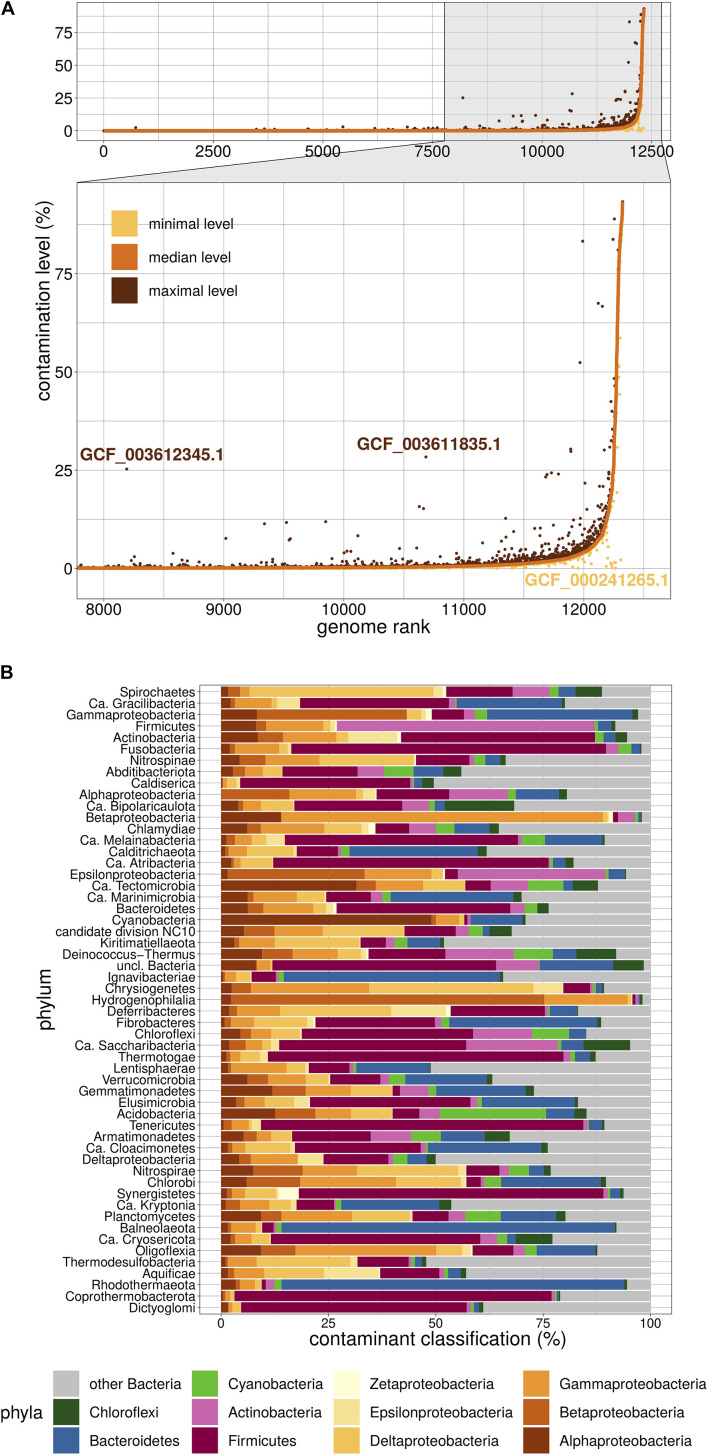
Contaminating sequences in public genome databases is a pervasive issue with potentially far-reaching consequences. This problem has attracted much attention in the recent literature and many different tools are now available to detect contaminants. Although these methods are based on diverse algorithms that can sometimes produce widely different estimates of the contamination level, the majority of genomic studies rely on a single method of detection, which represents a risk of systematic error. In this work, we used two orthogonal methods to assess the level of contamination among National Center for Biotechnological Information Reference Sequence Database (RefSeq) bacterial genomes. First, we applied the most popular solution, CheckM, which is based on gene markers. We then complemented this approach by a genome-wide method, termed Physeter, which now implements a k-folds algorithm to avoid inaccurate detection due to potential contamination of the reference database. We demonstrate that CheckM cannot currently be applied to all available genomes and bacterial groups. While it performed well on the majority of RefSeq genomes, it produced dubious results for 12,326 organisms. Among those, Physeter identified 239 contaminated genomes that had been missed by CheckM. In conclusion, we emphasize the importance of using multiple methods of detection while providing an upgrade of our own detection tool, Physeter, which minimizes incorrect contamination estimates in the context of unavoidably contaminated reference databases.
Keywords: NCBI RefSeq; assembly; contamination; databases; genomes; phylogenomics; sequencing.
Copyright © 2021 Lupo, Van Vlierberghe, Vanderschuren, Kerff, Baurain and Cornet.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Associated data
LinkOut - more resources
Full Text Sources