

Citrate Promotes Excessive Lipid Biosynthesis and Senescence in Tumor Cells for Tumor Therapy
- PMID: 34747157
- PMCID: PMC8728847
- DOI: 10.1002/advs.202101553
Citrate Promotes Excessive Lipid Biosynthesis and Senescence in Tumor Cells for Tumor Therapy
Abstract
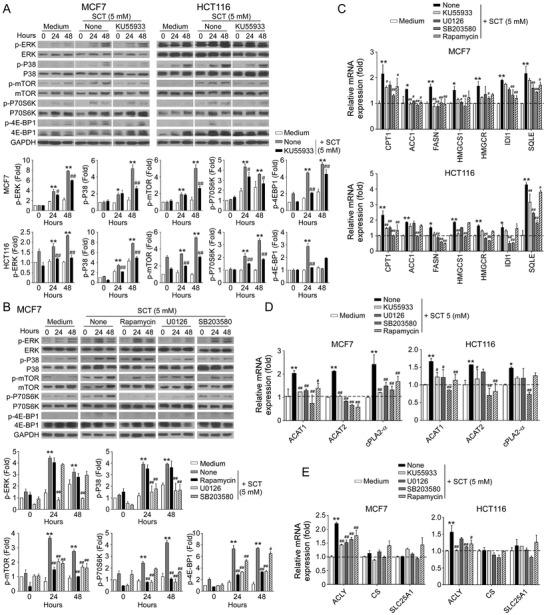
Metabolic disorder is one of the hallmarks of cancers, and reprogramming of metabolism is becoming a novel strategy for cancer treatment. Citrate is a key metabolite and critical metabolic regulator linking glycolysis and lipid metabolism in cellular energy homeostasis. Here it is reported that citrate treatment (both sodium citrate and citric acid) significantly suppresses tumor cell proliferation and growth in various tumor types. Mechanistically, citrate promotes excessive lipid biosynthesis and induces disruption of lipid metabolism in tumor cells, resulting in tumor cell senescence and growth inhibition. Furthermore, ATM-associated DNA damage response cooperates with MAPK and mTOR signaling pathways to control citrate-induced tumor cell growth arrest and senescence. In vivo studies further demonstrate that citrate administration dramatically inhibits tumor growth and progression in a colon cancer xenograft model. Importantly, citrate administration combined with the conventional chemotherapy drugs exhibits synergistic antitumor effects in vivo in the colon cancer models. These results clearly indicate that citrate can reprogram lipid metabolism and cell fate in cancer cells, and targeting citrate can be a promising therapeutic strategy for tumor treatment.
Keywords: DNA damage response; MAPK; apoptosis; cellular senescence; chemotherapy; citrate; lipid metabolism; mTOR.
© 2021 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








References
-
- Martinez‐Outschoorn U. E., Peiris‐Pages M., Pestell R. G., Sotgia F., Lisanti M. P., Nat. Rev. Clin. Oncol. 2017, 14, 11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous