

Metaproteomics reveals insights into microbial structure, interactions, and dynamic regulation in defined communities as they respond to environmental disturbance
- PMID: 34749649
- PMCID: PMC8574000
- DOI: 10.1186/s12866-021-02370-4
Metaproteomics reveals insights into microbial structure, interactions, and dynamic regulation in defined communities as they respond to environmental disturbance
Abstract
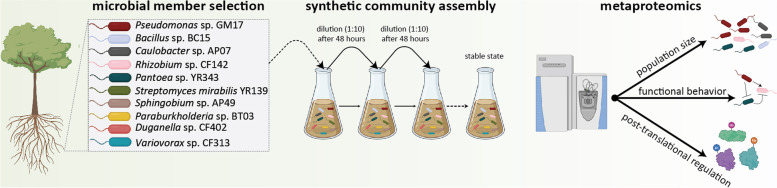
Background: Microbe-microbe interactions between members of the plant rhizosphere are important but remain poorly understood. A more comprehensive understanding of the molecular mechanisms used by microbes to cooperate, compete, and persist has been challenging because of the complexity of natural ecosystems and the limited control over environmental factors. One strategy to address this challenge relies on studying complexity in a progressive manner, by first building a detailed understanding of relatively simple subsets of the community and then achieving high predictive power through combining different building blocks (e.g., hosts, community members) for different environments. Herein, we coupled this reductionist approach with high-resolution mass spectrometry-based metaproteomics to study molecular mechanisms driving community assembly, adaptation, and functionality for a defined community of ten taxonomically diverse bacterial members of Populus deltoides rhizosphere co-cultured either in a complex or defined medium.
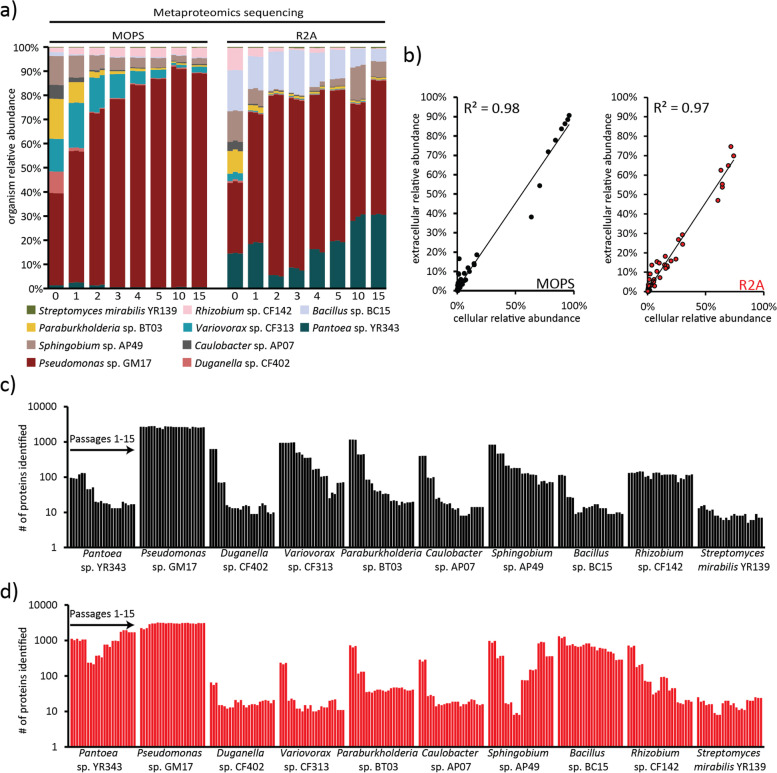
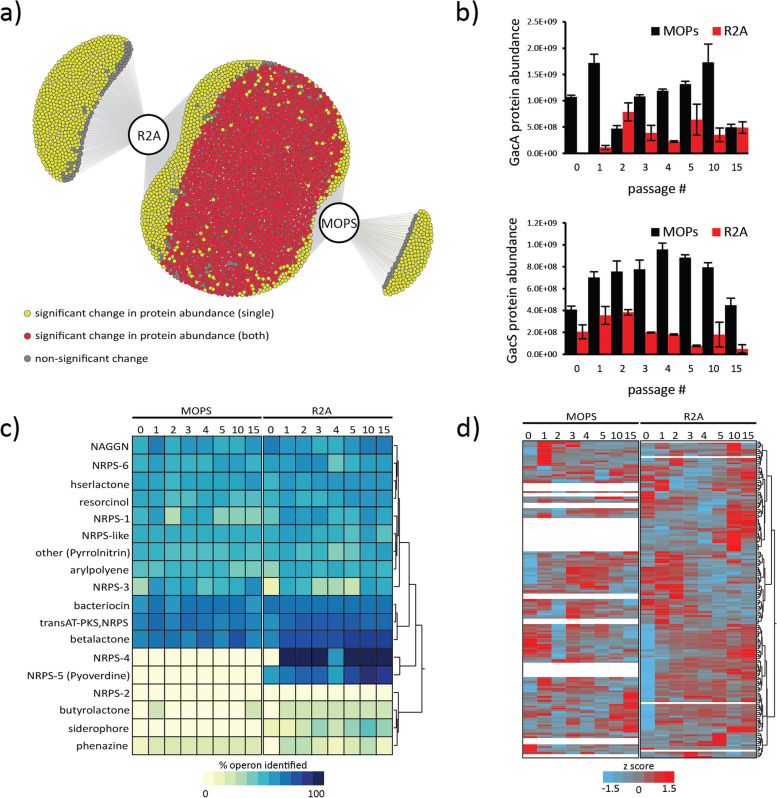
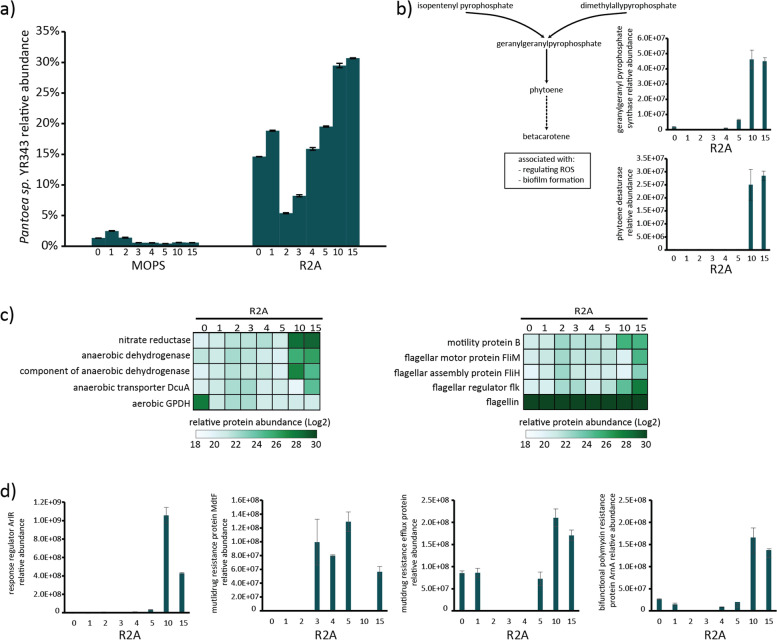
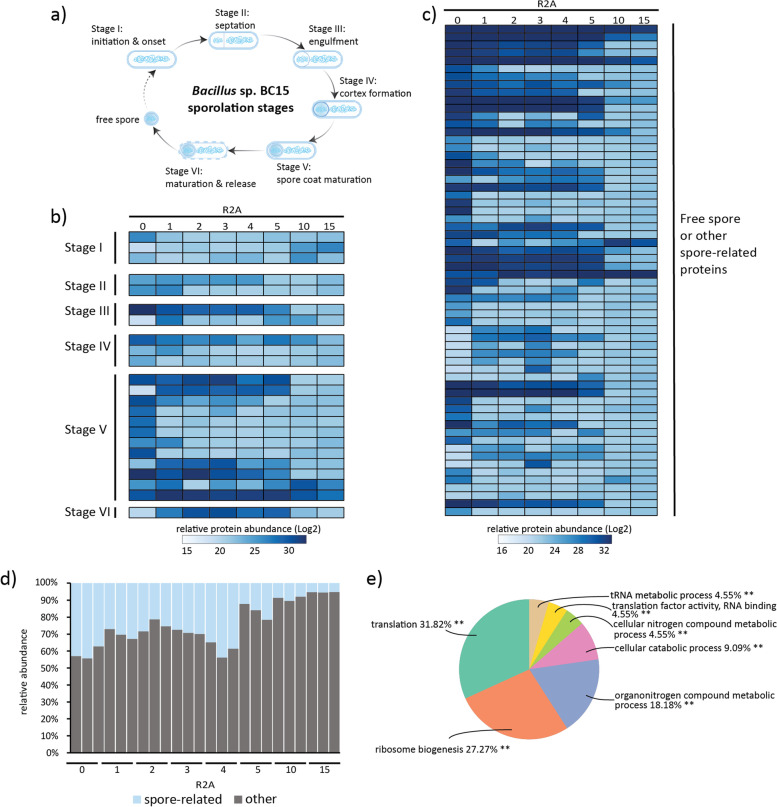
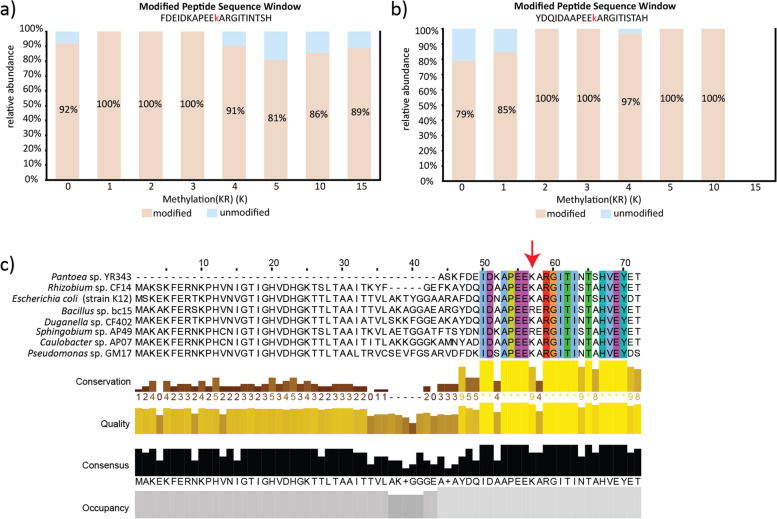
Results: Metaproteomics showed this defined community assembled into distinct microbiomes based on growth media that eventually exhibit composition and functional stability over time. The community grown in two different media showed variation in composition, yet both were dominated by only a few microbial strains. Proteome-wide interrogation provided detailed insights into the functional behavior of each dominant member as they adjust to changing community compositions and environments. The emergence and persistence of select microbes in these communities were driven by specialization in strategies including motility, antibiotic production, altered metabolism, and dormancy. Protein-level interrogation identified post-translational modifications that provided additional insights into regulatory mechanisms influencing microbial adaptation in the changing environments.
Conclusions: This study provides high-resolution proteome-level insights into our understanding of microbe-microbe interactions and highlights specialized biological processes carried out by specific members of assembled microbiomes to compete and persist in changing environmental conditions. Emergent properties observed in these lower complexity communities can then be re-evaluated as more complex systems are studied and, when a particular property becomes less relevant, higher-order interactions can be identified.
Keywords: Defined community; Metaproteomics; Microbial consortia; PTMs; Reductionist approach; Rhizospheric microbiome.
© 2021. The Author(s).
Conflict of interest statement
There is no potential conflict of interests.
Figures
References
-
- Pii Y, Mimmo T, Tomasi N, Terzano R, Cesco S, Crecchio C, Microbial interactions in the rhizosphere: beneficial influences of plant growth-promoting rhizobacteria on nutrient acquisition process. Biol Fertility Soils. 2015; 51(4):403-415.
-
- De Zelicourt A, Al-Yousif M, Hirt H. Rhizosphere microbes as essential partners for plant stress tolerance. Molecular Plant. 2013;6(2):242–245. - PubMed
-
- Berendsen RL, Pieterse CM, Bakker PA. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012;17(8):478–86. - PubMed
-
- Kumar M, et al. Modelling approaches for studying the microbiome. Nat Microbiol. 2019;4(8):1253–1267. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
