

Integrative analysis of the plasma proteome and polygenic risk of cardiometabolic diseases
- PMID: 34750571
- PMCID: PMC8574944
- DOI: 10.1038/s42255-021-00478-5
Integrative analysis of the plasma proteome and polygenic risk of cardiometabolic diseases
Abstract
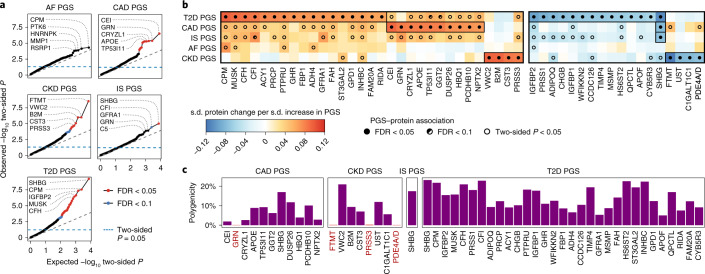
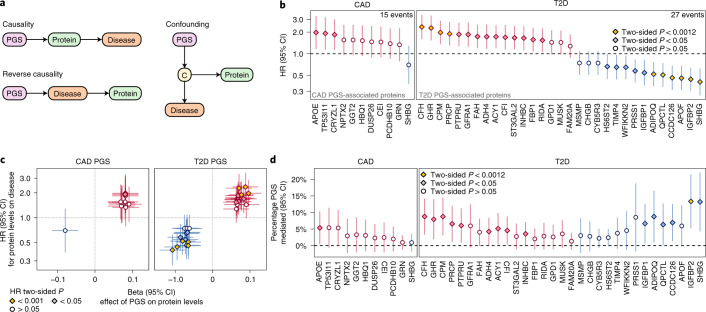
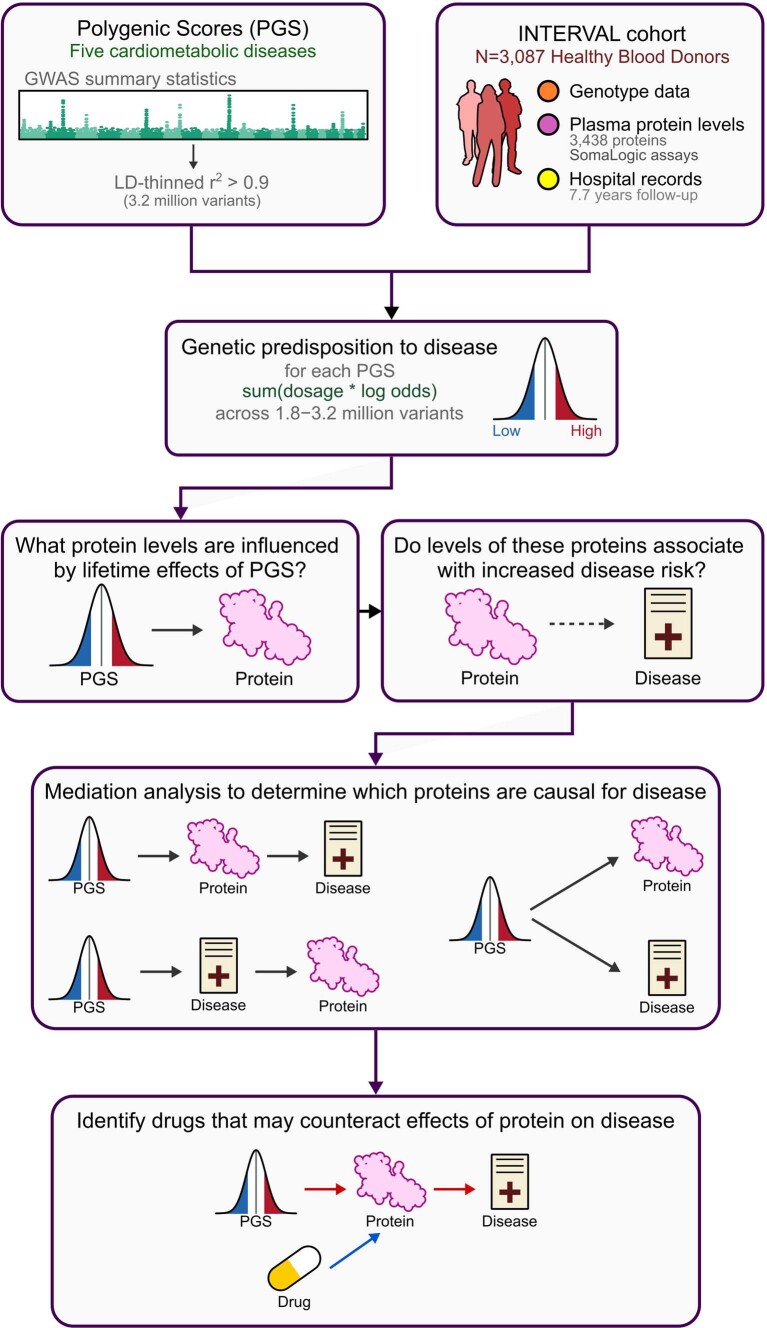
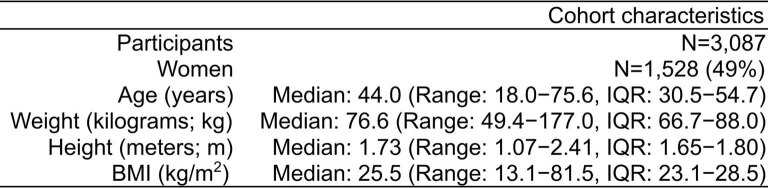
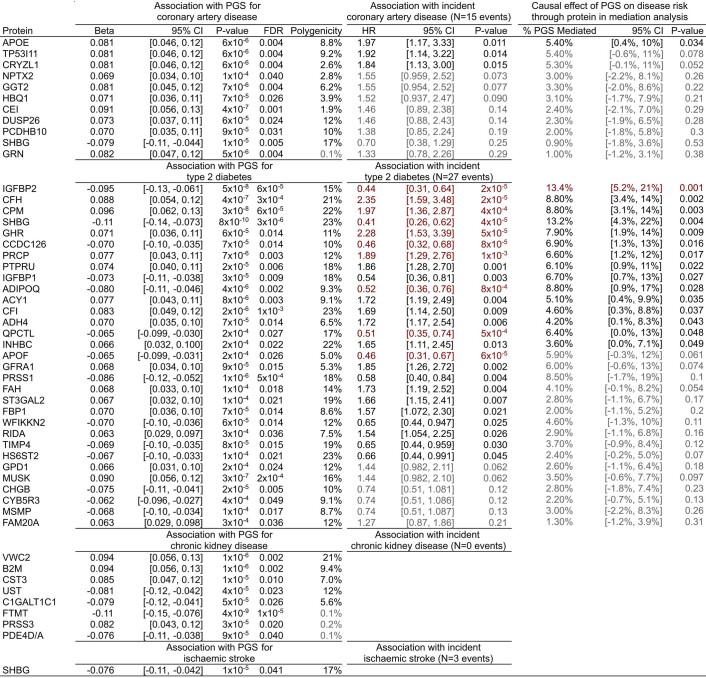
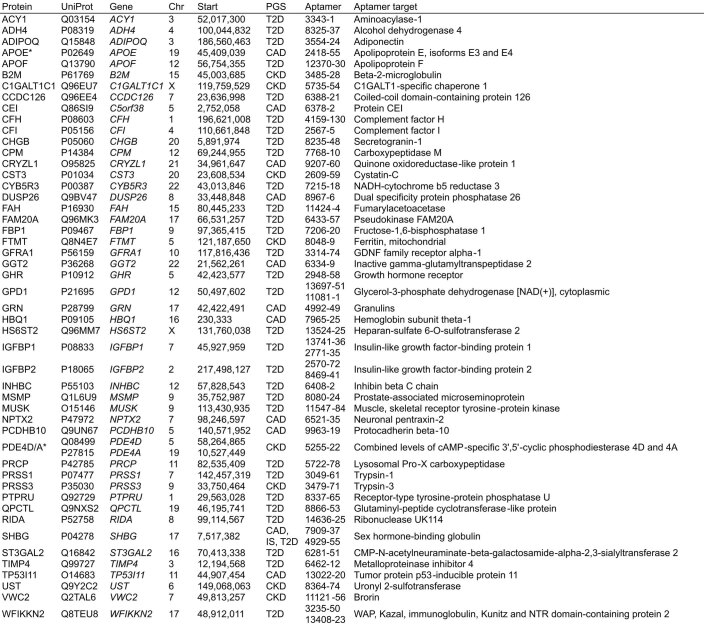
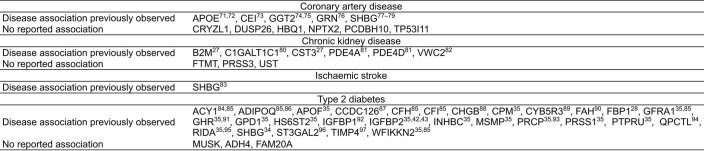
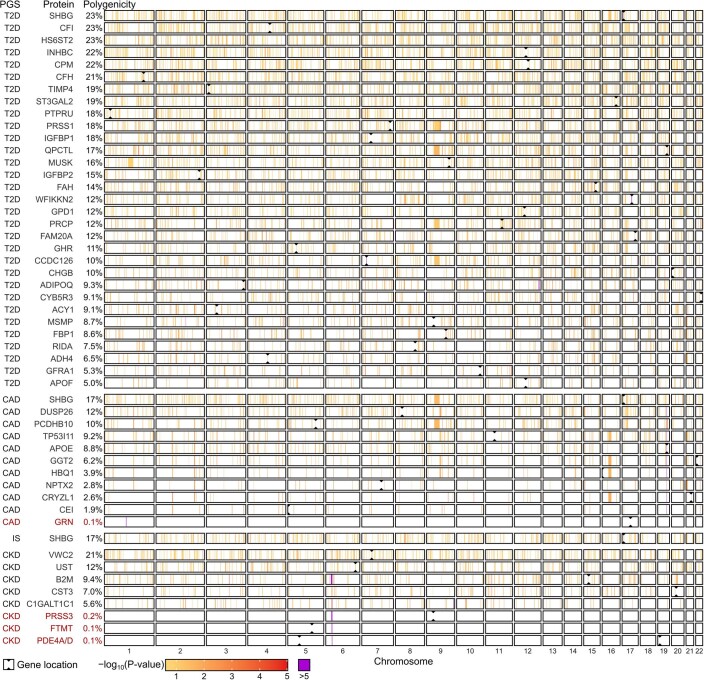
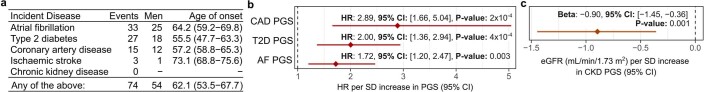
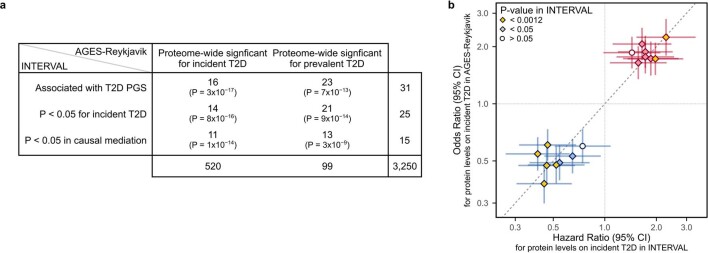
Cardiometabolic diseases are frequently polygenic in architecture, comprising a large number of risk alleles with small effects spread across the genome1-3. Polygenic scores (PGS) aggregate these into a metric representing an individual's genetic predisposition to disease. PGS have shown promise for early risk prediction4-7 and there is an open question as to whether PGS can also be used to understand disease biology8. Here, we demonstrate that cardiometabolic disease PGS can be used to elucidate the proteins underlying disease pathogenesis. In 3,087 healthy individuals, we found that PGS for coronary artery disease, type 2 diabetes, chronic kidney disease and ischaemic stroke are associated with the levels of 49 plasma proteins. Associations were polygenic in architecture, largely independent of cis and trans protein quantitative trait loci and present for proteins without quantitative trait loci. Over a follow-up of 7.7 years, 28 of these proteins associated with future myocardial infarction or type 2 diabetes events, 16 of which were mediators between polygenic risk and incident disease. Twelve of these were druggable targets with therapeutic potential. Our results demonstrate the potential for PGS to uncover causal disease biology and targets with therapeutic potential, including those that may be missed by approaches utilizing information at a single locus.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Several authors are now employed by or run pharmaceutical companies. All significant contributions to this study were made before these roles and the named companies had no role in the study. M.A. is an employee of AstraZeneca. P.S. is an employee of Roche. J.M. is an employee of Genomics PLC. G.A. is an employee of CSL Limited. S.K. is the chief executive officer of Verve Therapeutics. The other authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
