

Mechanisms and Treatments in Demyelinating CMT
- PMID: 34750751
- PMCID: PMC8804145
- DOI: 10.1007/s13311-021-01145-z
Mechanisms and Treatments in Demyelinating CMT
Abstract
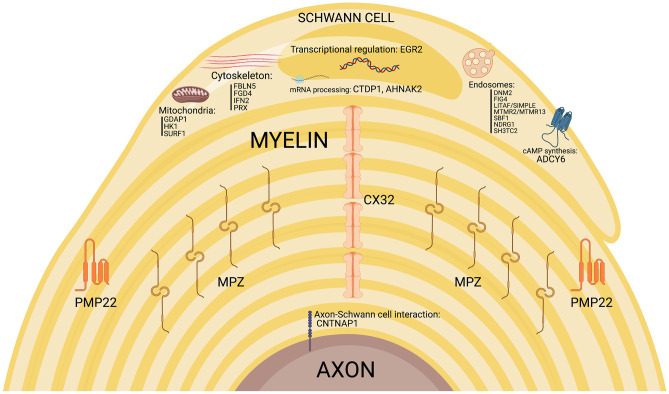
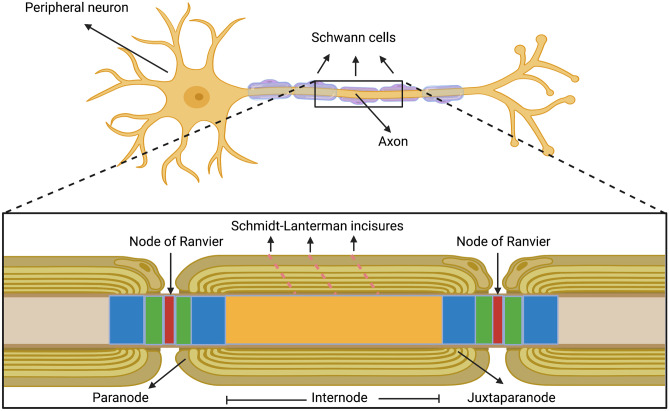
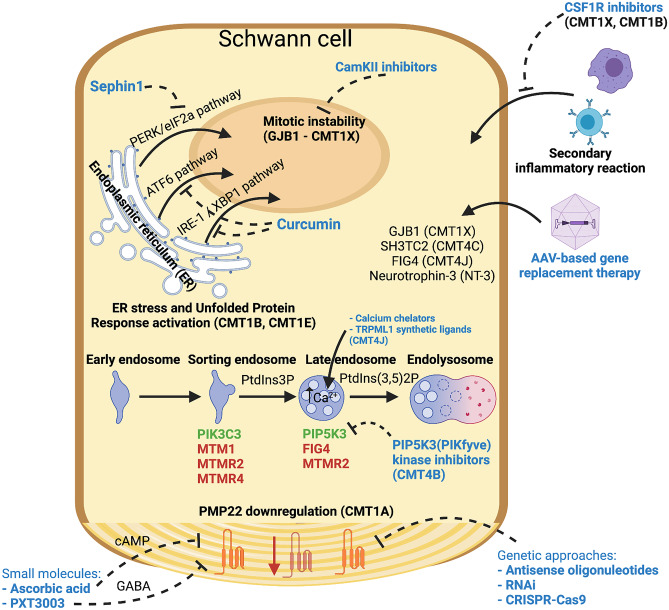
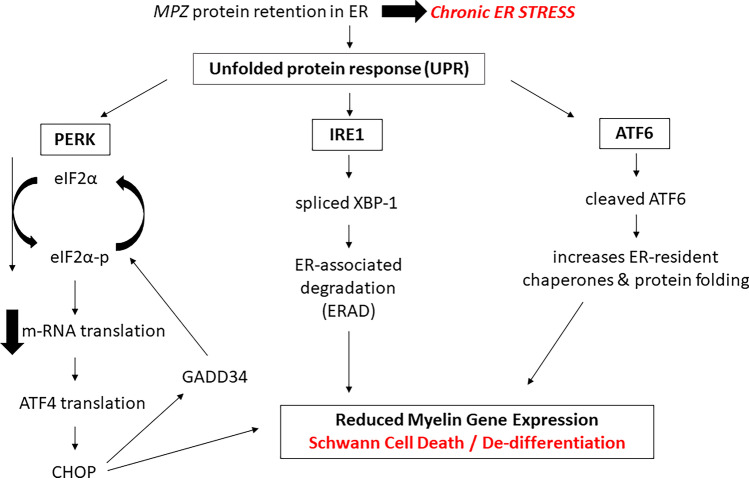
Demyelinating forms of Charcot-Marie-Tooth disease (CMT) are genetically and phenotypically heterogeneous and result from highly diverse biological mechanisms including gain of function (including dominant negative effects) and loss of function. While no definitive treatment is currently available, rapid advances in defining the pathomechanisms of demyelinating CMT have led to promising pre-clinical studies, as well as emerging clinical trials. Especially promising are the recently completed pre-clinical genetic therapy studies in PMP-22, GJB1, and SH3TC2-associated neuropathies, particularly given the success of similar approaches in humans with spinal muscular atrophy and transthyretin familial polyneuropathy. This article focuses on neuropathies related to mutations in PMP-22, MPZ, and GJB1, which together comprise the most common forms of demyelinating CMT, as well as on select rarer forms for which promising treatment targets have been identified. Clinical characteristics and pathomechanisms are reviewed in detail, with emphasis on therapeutically targetable biological pathways. Also discussed are the challenges facing the CMT research community in its efforts to advance the rapidly evolving biological insights to effective clinical trials. These considerations include the limitations of currently available animal models, the need for personalized medicine approaches/allele-specific interventions for select forms of demyelinating CMT, and the increasing demand for optimal clinical outcome assessments and objective biomarkers.
Keywords: Biological mechanisms; Charcot-Marie-Tooth disease; Clinical trials; Demyelinating neuropathy; Therapeutic development; Treatment targets.
© 2021. The American Society for Experimental NeuroTherapeutics, Inc.
Figures
References
-
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6(2):98–118. - PubMed
-
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259–280. - PubMed
-
- Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM. Updating the classification of inherited neuropathies: Results of an international survey. Neurology. 2018;90(10):e870–e876. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
