

Modeling Sequence-Space Exploration and Emergence of Epistatic Signals in Protein Evolution
- PMID: 34751386
- PMCID: PMC8789065
- DOI: 10.1093/molbev/msab321
Modeling Sequence-Space Exploration and Emergence of Epistatic Signals in Protein Evolution
Abstract
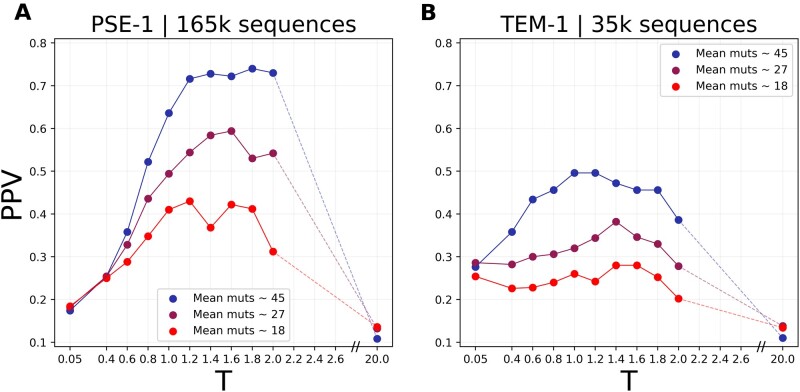
During their evolution, proteins explore sequence space via an interplay between random mutations and phenotypic selection. Here, we build upon recent progress in reconstructing data-driven fitness landscapes for families of homologous proteins, to propose stochastic models of experimental protein evolution. These models predict quantitatively important features of experimentally evolved sequence libraries, like fitness distributions and position-specific mutational spectra. They also allow us to efficiently simulate sequence libraries for a vast array of combinations of experimental parameters like sequence divergence, selection strength, and library size. We showcase the potential of the approach in reanalyzing two recent experiments to determine protein structure from signals of epistasis emerging in experimental sequence libraries. To be detectable, these signals require sufficiently large and sufficiently diverged libraries. Our modeling framework offers a quantitative explanation for different outcomes of recently published experiments. Furthermore, we can forecast the outcome of time- and resource-intensive evolution experiments, opening thereby a way to computationally optimize experimental protocols.
Keywords: data-driven models; epistasis; fitness landscapes; protein evolution; sequence space.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures





References
-
- Ackley DH, Hinton GE, Sejnowski TJ.. 1985. A learning algorithm for Boltzmann machines. Cogn Sci. 9(1):147–169.
-
- Arnold FH. 1998. Design by directed evolution. Acc Chem Res. 31(3):125–131.
-
- Balakrishnan S, Kamisetty H, Carbonell JG, Lee S-I, Langmead CJ.. 2011. Learning generative models for protein fold families. Proteins 79(4):1061–1078. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
