Diversity, taxonomy, and evolution of archaeal viruses of the class Caudoviricetes
- PMID: 34752450
- PMCID: PMC8651126
- DOI: 10.1371/journal.pbio.3001442
Diversity, taxonomy, and evolution of archaeal viruses of the class Caudoviricetes
Abstract
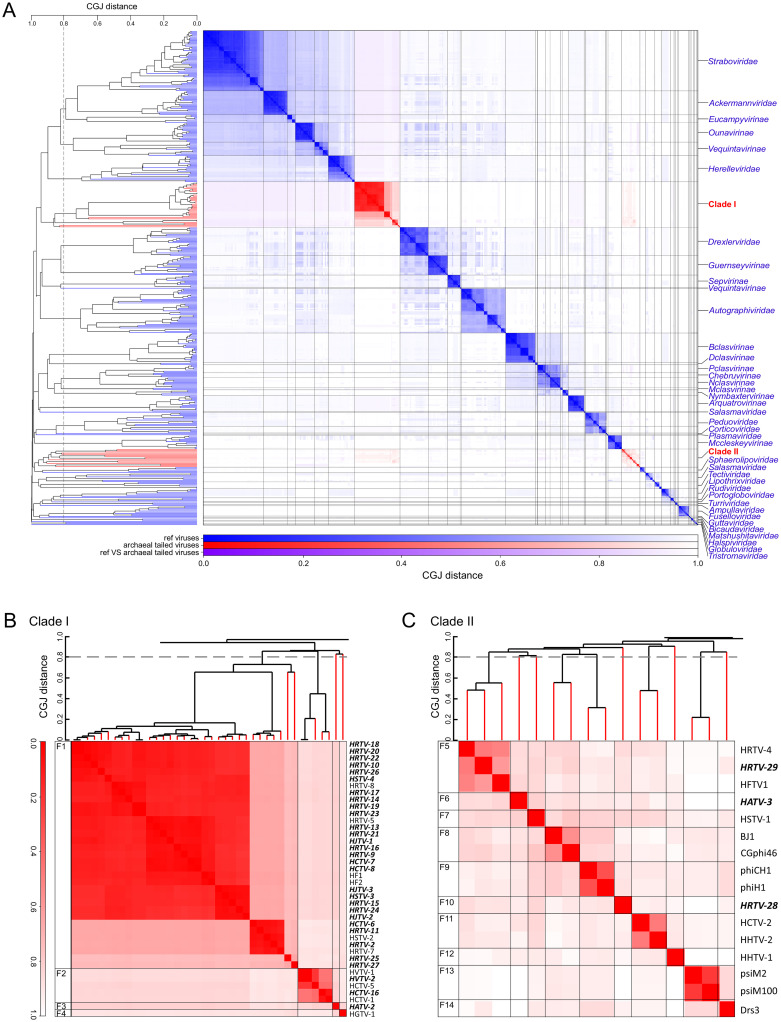
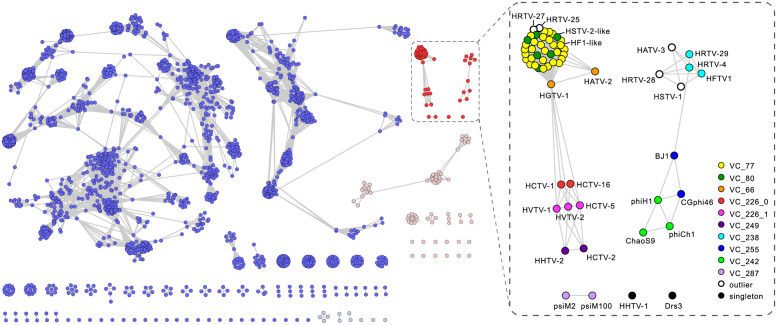
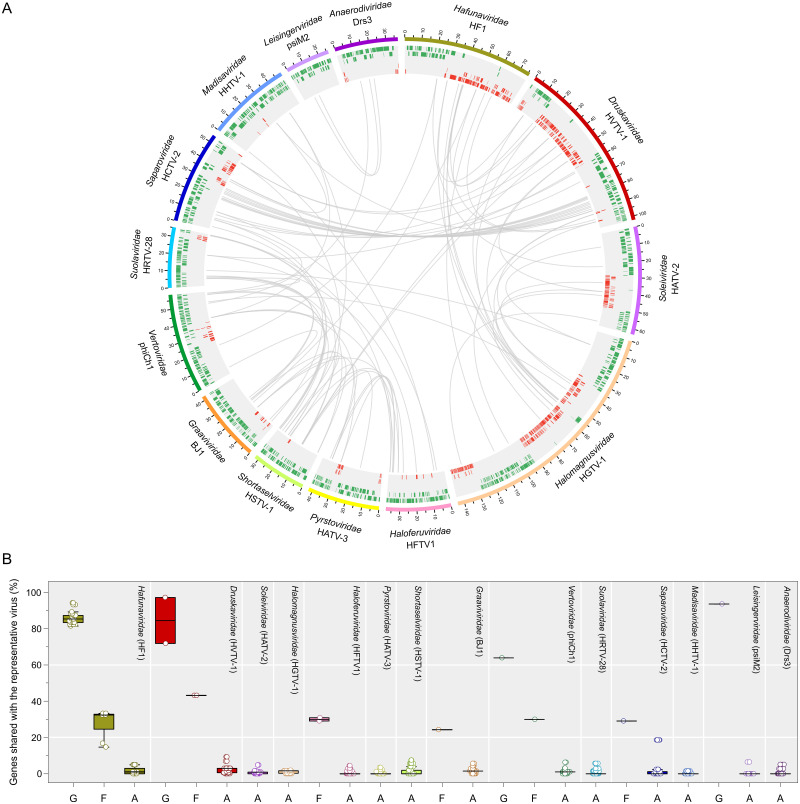
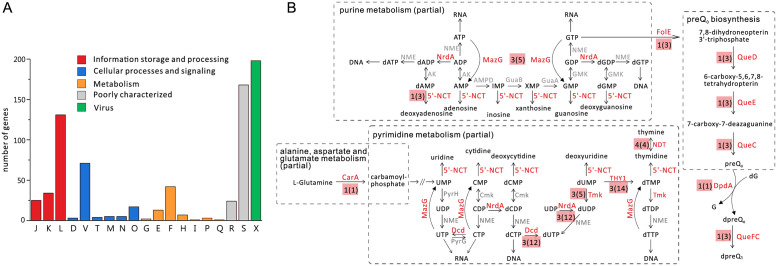
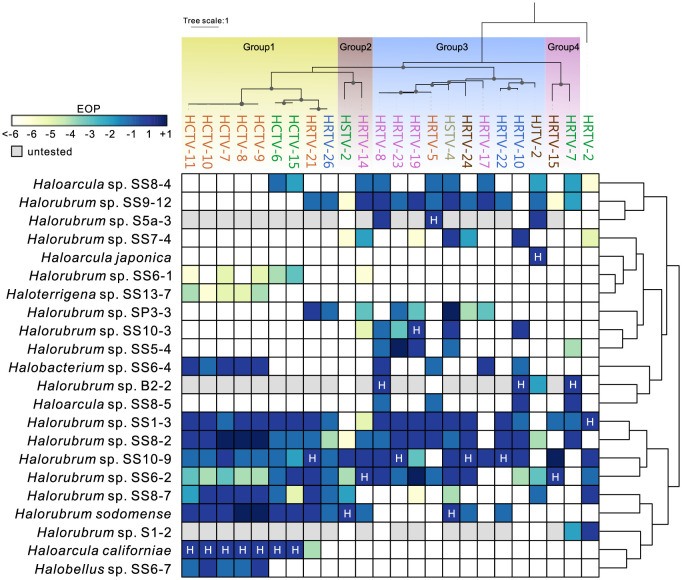
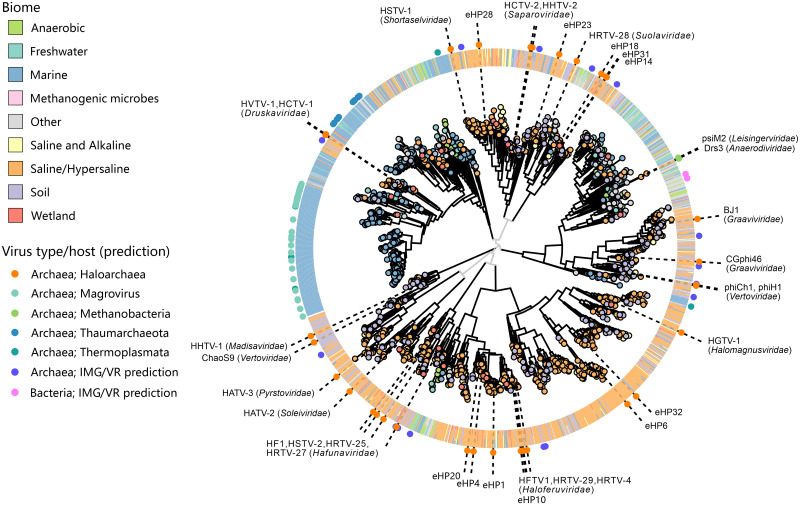
The archaeal tailed viruses (arTV), evolutionarily related to tailed double-stranded DNA (dsDNA) bacteriophages of the class Caudoviricetes, represent the most common isolates infecting halophilic archaea. Only a handful of these viruses have been genomically characterized, limiting our appreciation of their ecological impacts and evolution. Here, we present 37 new genomes of haloarchaeal tailed virus isolates, more than doubling the current number of sequenced arTVs. Analysis of all 63 available complete genomes of arTVs, which we propose to classify into 14 new families and 3 orders, suggests ancient divergence of archaeal and bacterial tailed viruses and points to an extensive sharing of genes involved in DNA metabolism and counterdefense mechanisms, illuminating common strategies of virus-host interactions with tailed bacteriophages. Coupling of the comparative genomics with the host range analysis on a broad panel of haloarchaeal species uncovered 4 distinct groups of viral tail fiber adhesins controlling the host range expansion. The survey of metagenomes using viral hallmark genes suggests that the global architecture of the arTV community is shaped through recurrent transfers between different biomes, including hypersaline, marine, and anoxic environments.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures