

Whole genome and exome sequencing reference datasets from a multi-center and cross-platform benchmark study
- PMID: 34753956
- PMCID: PMC8578599
- DOI: 10.1038/s41597-021-01077-5
Whole genome and exome sequencing reference datasets from a multi-center and cross-platform benchmark study
Abstract
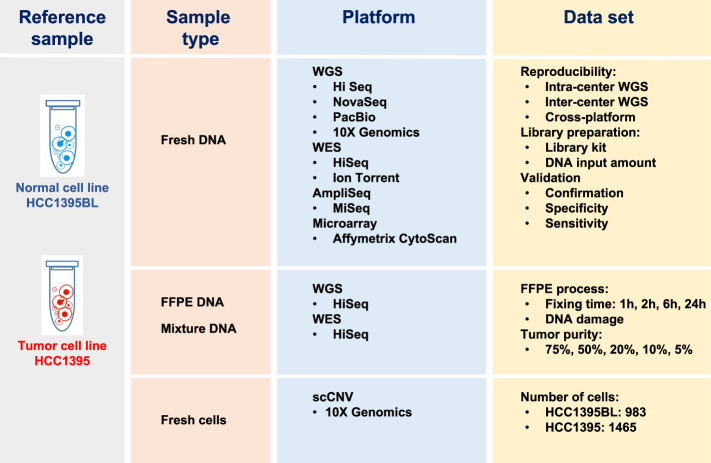
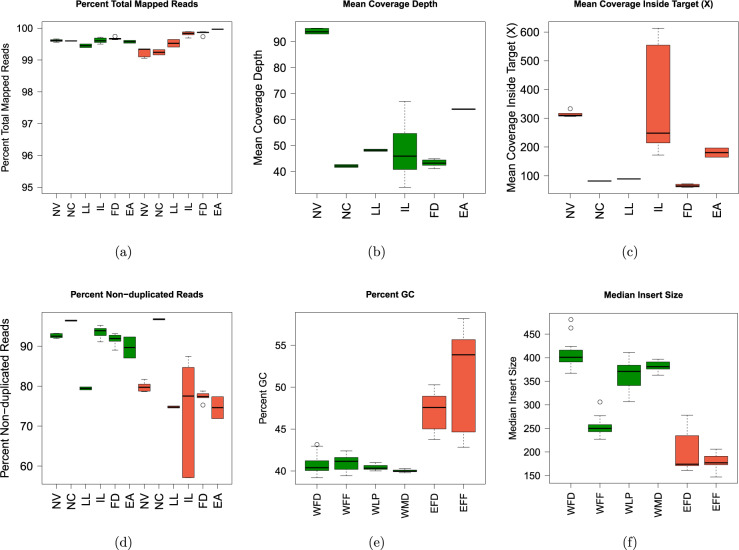
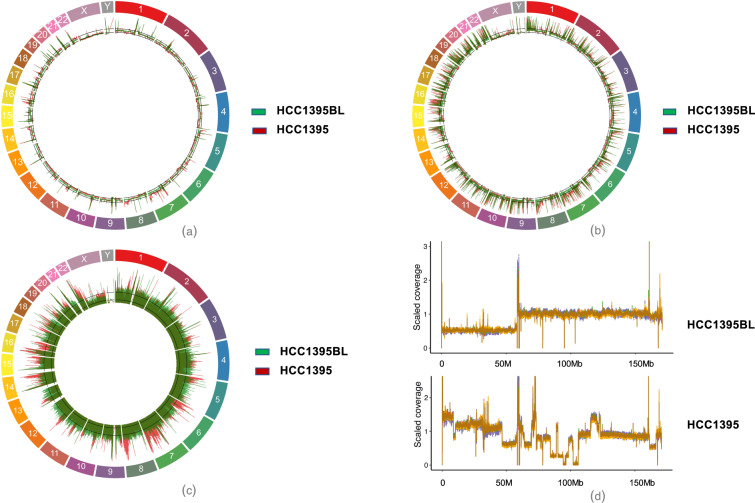
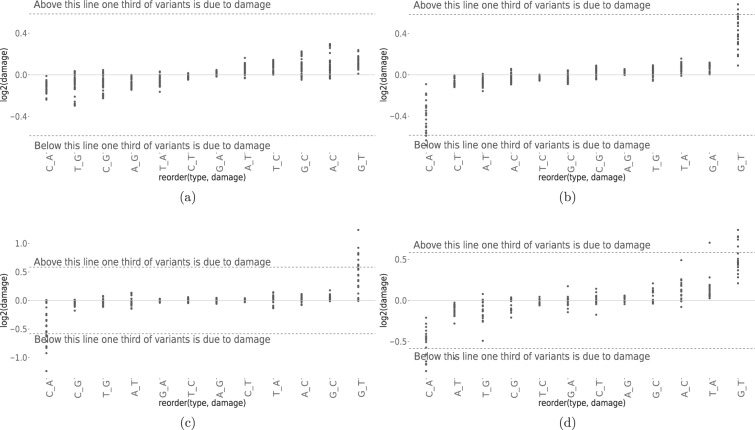
With the rapid advancement of sequencing technologies, next generation sequencing (NGS) analysis has been widely applied in cancer genomics research. More recently, NGS has been adopted in clinical oncology to advance personalized medicine. Clinical applications of precision oncology require accurate tests that can distinguish tumor-specific mutations from artifacts introduced during NGS processes or data analysis. Therefore, there is an urgent need to develop best practices in cancer mutation detection using NGS and the need for standard reference data sets for systematically measuring accuracy and reproducibility across platforms and methods. Within the SEQC2 consortium context, we established paired tumor-normal reference samples and generated whole-genome (WGS) and whole-exome sequencing (WES) data using sixteen library protocols, seven sequencing platforms at six different centers. We systematically interrogated somatic mutations in the reference samples to identify factors affecting detection reproducibility and accuracy in cancer genomes. These large cross-platform/site WGS and WES datasets using well-characterized reference samples will represent a powerful resource for benchmarking NGS technologies, bioinformatics pipelines, and for the cancer genomics studies.
© 2021. The Author(s).
Conflict of interest statement
Li Tai Fang is employee of Roche Sequencing Solutions Inc. Erich Jaeger is employee of Illumina Inc. Virginie Petitjean and Marc Sultan are employees of Novartis Institutes for Biomedical Research. Tiffany Hung and Eric Peters are employees of Genentech (a member of the Roche group). All other authors claim no conflicts of interest. This is a research study, not intended to guide clinical applications. The views presented in this article do not necessarily reflect current or future opinion or policy of the US Food and Drug Administration. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services. Any mention of commercial products is for clarification and not intended as endorsement.
Figures
Dataset use reported in
References
-
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv, https://arxiv.org/abs/1303.3997 (2013).
Publication types
MeSH terms
Grants and funding
- Project No. 2014-2020.4.01.15-0012/EC | European Regional Development Fund (Europski Fond za Regionalni Razvoj)
- HHSN261201500003C/CA/NCI NIH HHS/United States
- 2017-00630, 2019-01976/Vetenskapsrådet (Swedish Research Council)
- S10 OD019960/OD/NIH HHS/United States
- HHSN261201500003I/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
