

p38γ and p38δ regulate postnatal cardiac metabolism through glycogen synthase 1
- PMID: 34758018
- PMCID: PMC8612745
- DOI: 10.1371/journal.pbio.3001447
p38γ and p38δ regulate postnatal cardiac metabolism through glycogen synthase 1
Abstract
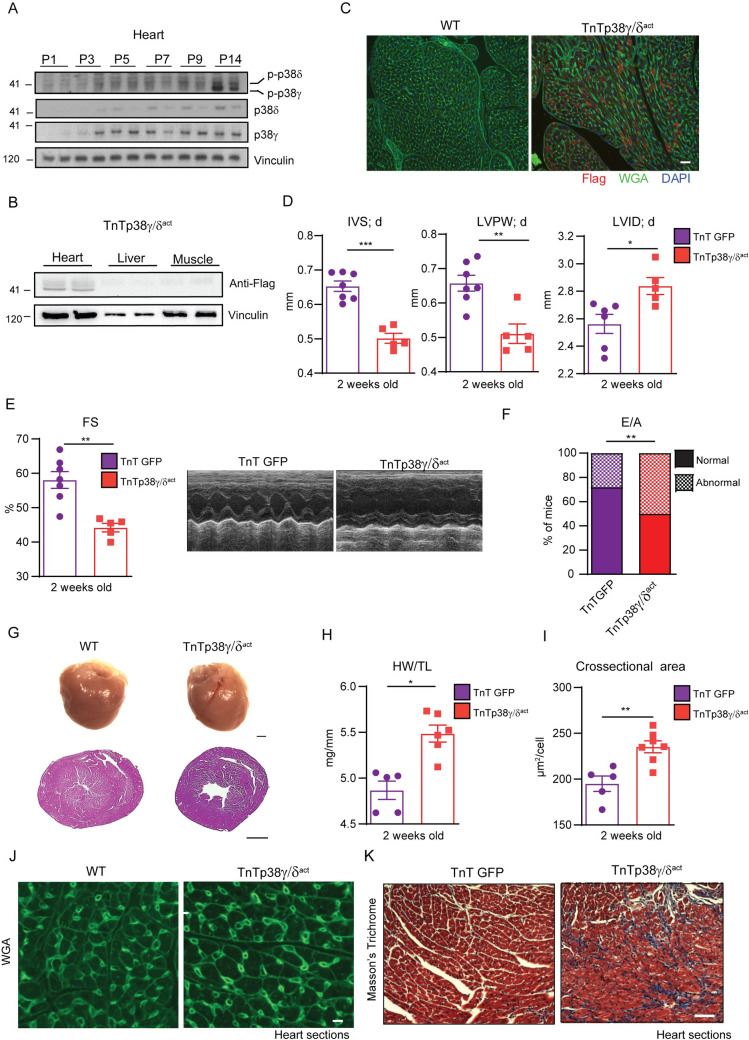
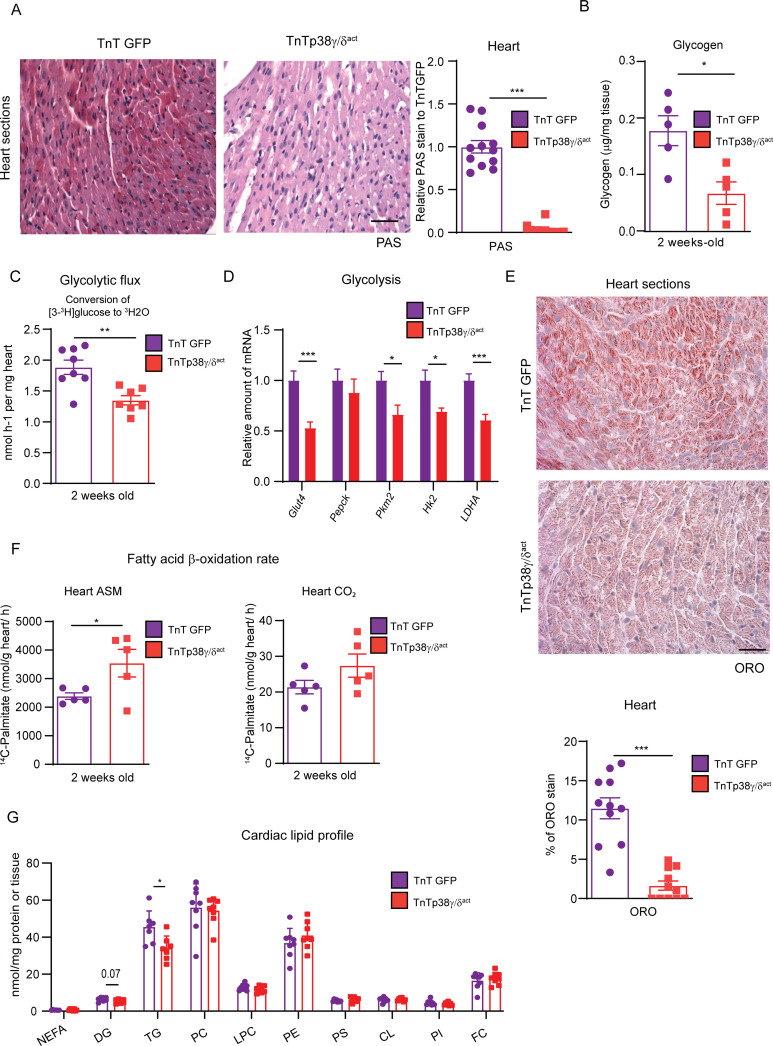
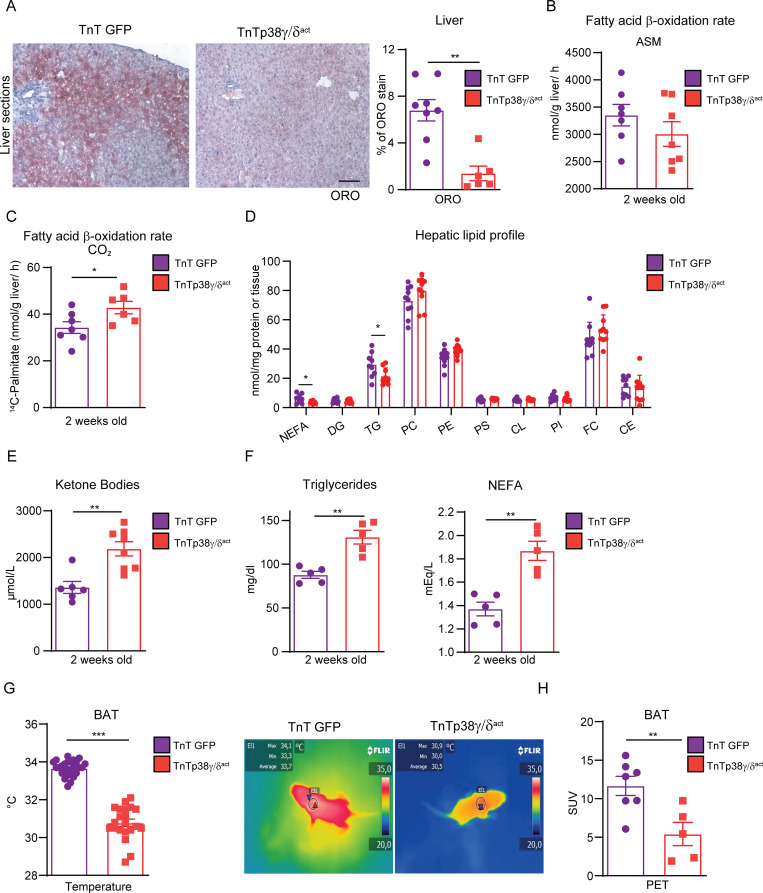
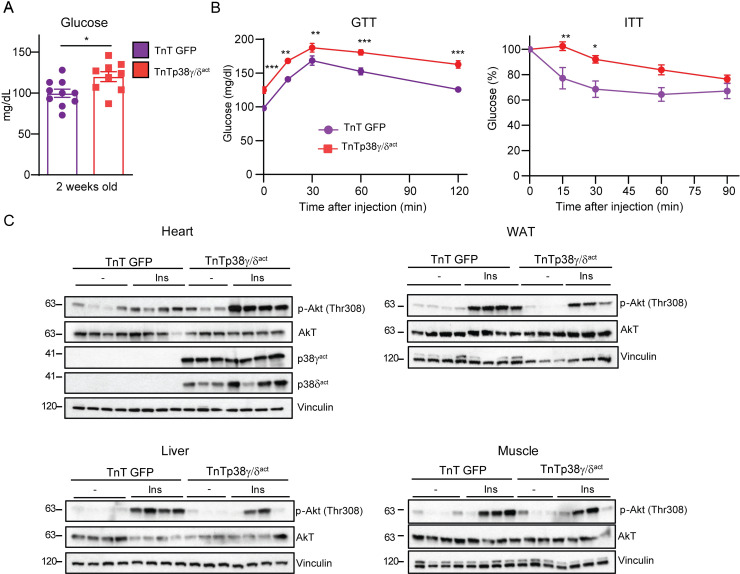
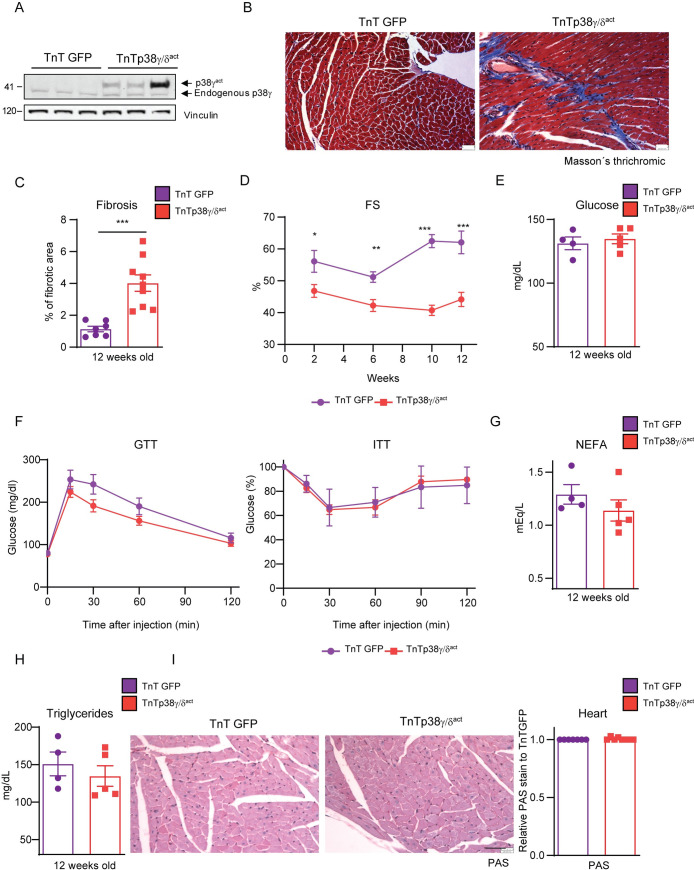
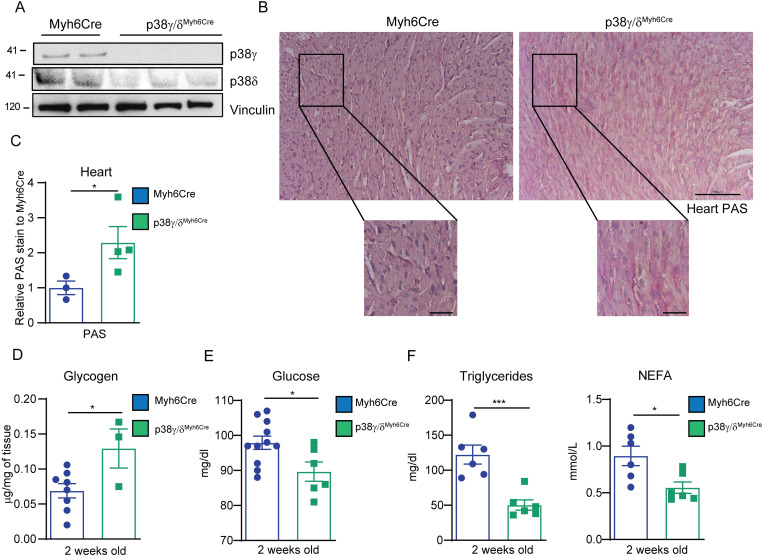
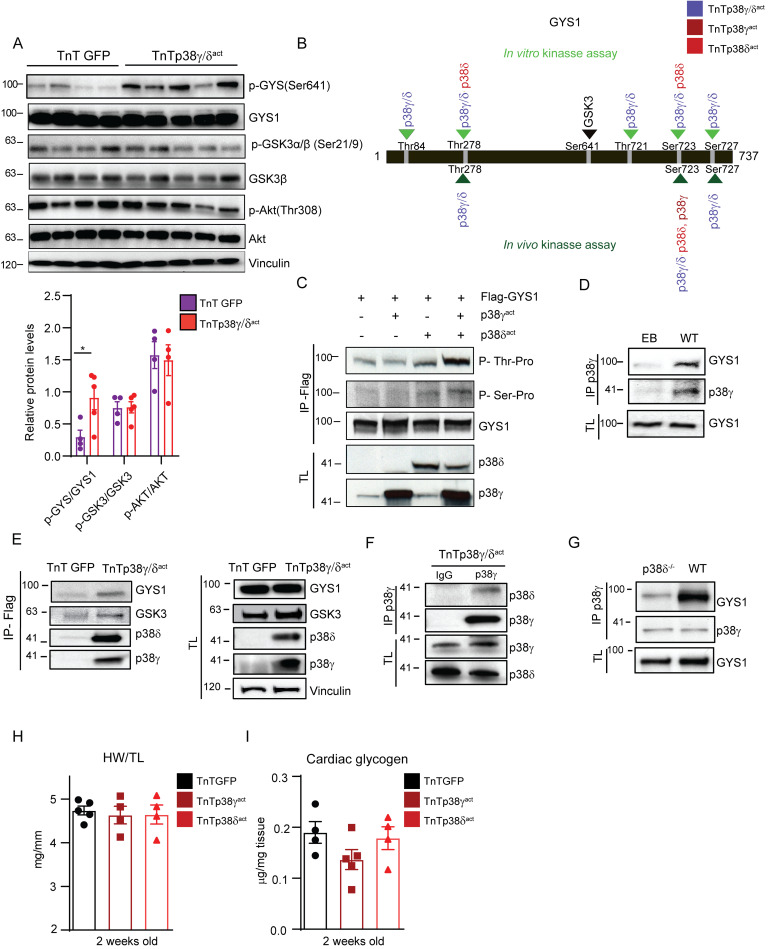
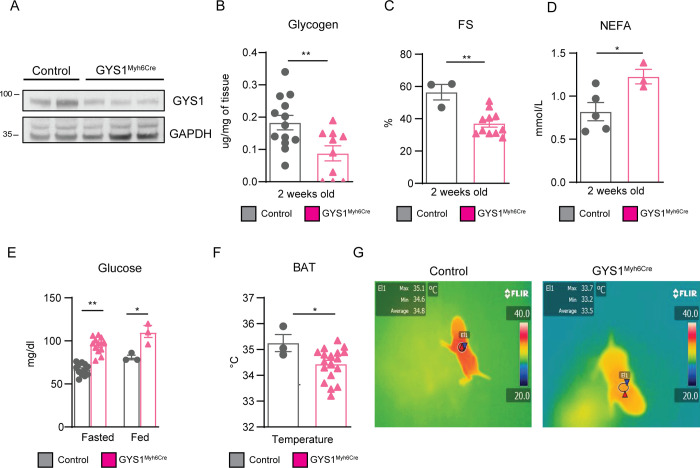
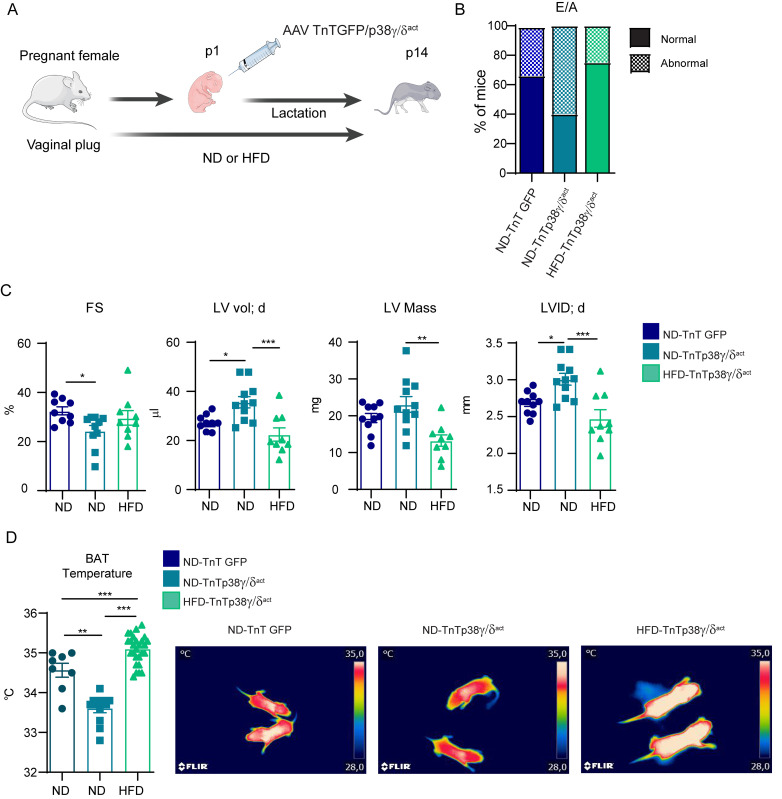
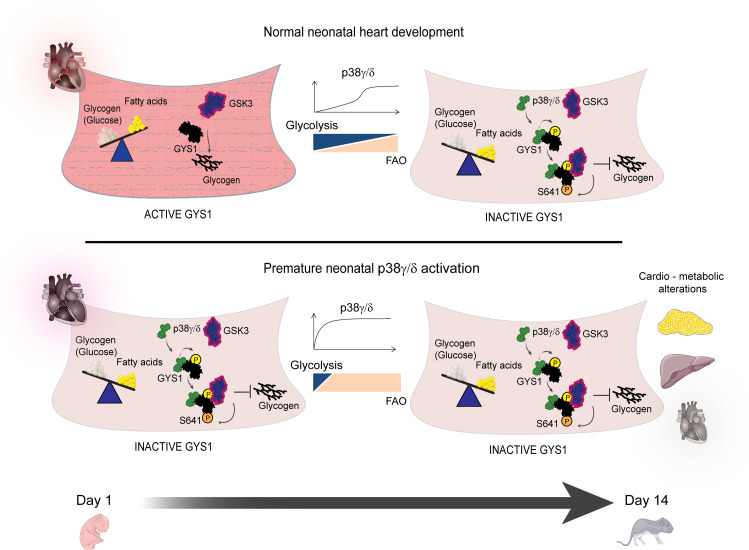
During the first weeks of postnatal heart development, cardiomyocytes undergo a major adaptive metabolic shift from glycolytic energy production to fatty acid oxidation. This metabolic change is contemporaneous to the up-regulation and activation of the p38γ and p38δ stress-activated protein kinases in the heart. We demonstrate that p38γ/δ contribute to the early postnatal cardiac metabolic switch through inhibitory phosphorylation of glycogen synthase 1 (GYS1) and glycogen metabolism inactivation. Premature induction of p38γ/δ activation in cardiomyocytes of newborn mice results in an early GYS1 phosphorylation and inhibition of cardiac glycogen production, triggering an early metabolic shift that induces a deficit in cardiomyocyte fuel supply, leading to whole-body metabolic deregulation and maladaptive cardiac pathogenesis. Notably, the adverse effects of forced premature cardiac p38γ/δ activation in neonate mice are prevented by maternal diet supplementation of fatty acids during pregnancy and lactation. These results suggest that diet interventions have a potential for treating human cardiac genetic diseases that affect heart metabolism.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med. 2007;4(Suppl 1):S60–7. Epub 2007 Jan 19. doi: 10.1038/ncpcardio0766 ; PubMed Central PMCID: PMC3232050. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
