

Crosstalk between the renin-angiotensin, complement and kallikrein-kinin systems in inflammation
- PMID: 34759348
- PMCID: PMC8579187
- DOI: 10.1038/s41577-021-00634-8
Crosstalk between the renin-angiotensin, complement and kallikrein-kinin systems in inflammation
Abstract
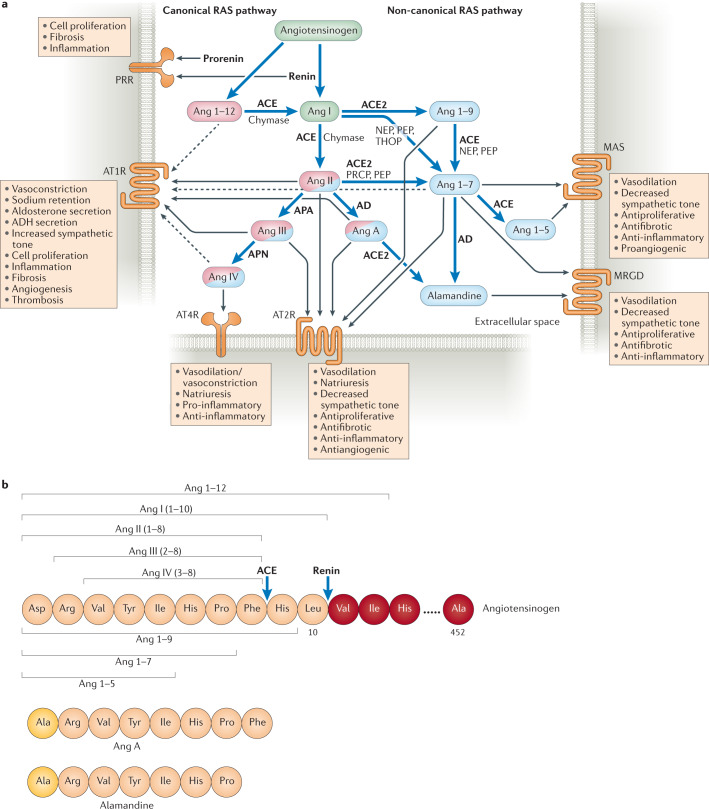
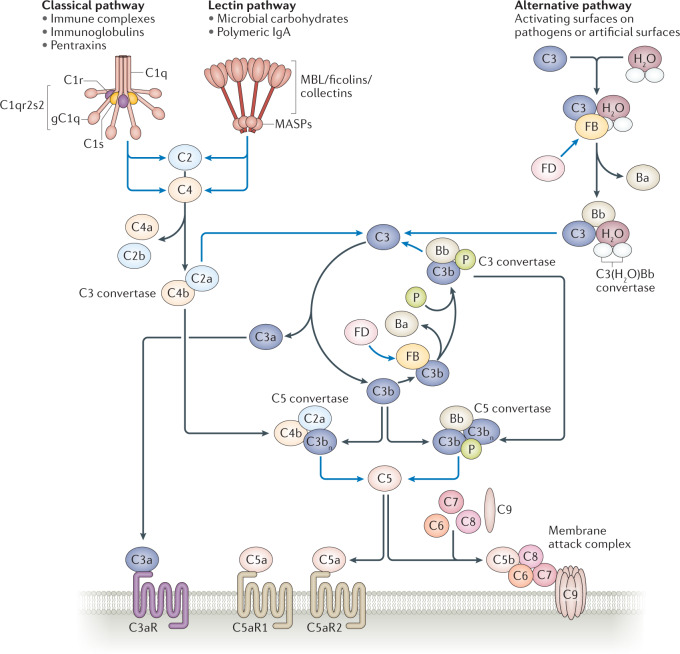
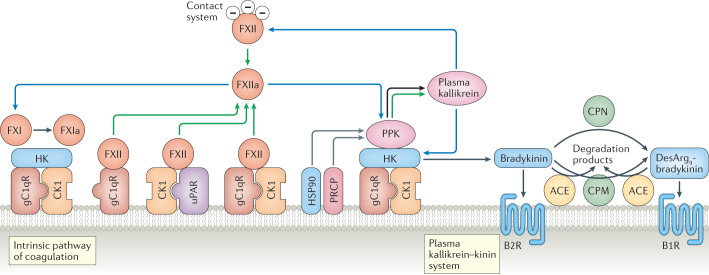
During severe inflammatory and infectious diseases, various mediators modulate the equilibrium of vascular tone, inflammation, coagulation and thrombosis. This Review describes the interactive roles of the renin-angiotensin system, the complement system, and the closely linked kallikrein-kinin and contact systems in cell biological functions such as vascular tone and leakage, inflammation, chemotaxis, thrombosis and cell proliferation. Specific attention is given to the role of these systems in systemic inflammation in the vasculature and tissues during hereditary angioedema, cardiovascular and renal glomerular disease, vasculitides and COVID-19. Moreover, we discuss the therapeutic implications of these complex interactions, given that modulation of one system may affect the other systems, with beneficial or deleterious consequences.
© 2021. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Colman RW, Schmaier AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–3843. - PubMed
-
- Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
