

Computed structures of core eukaryotic protein complexes
- PMID: 34762488
- PMCID: PMC7612107
- DOI: 10.1126/science.abm4805
Computed structures of core eukaryotic protein complexes
Abstract
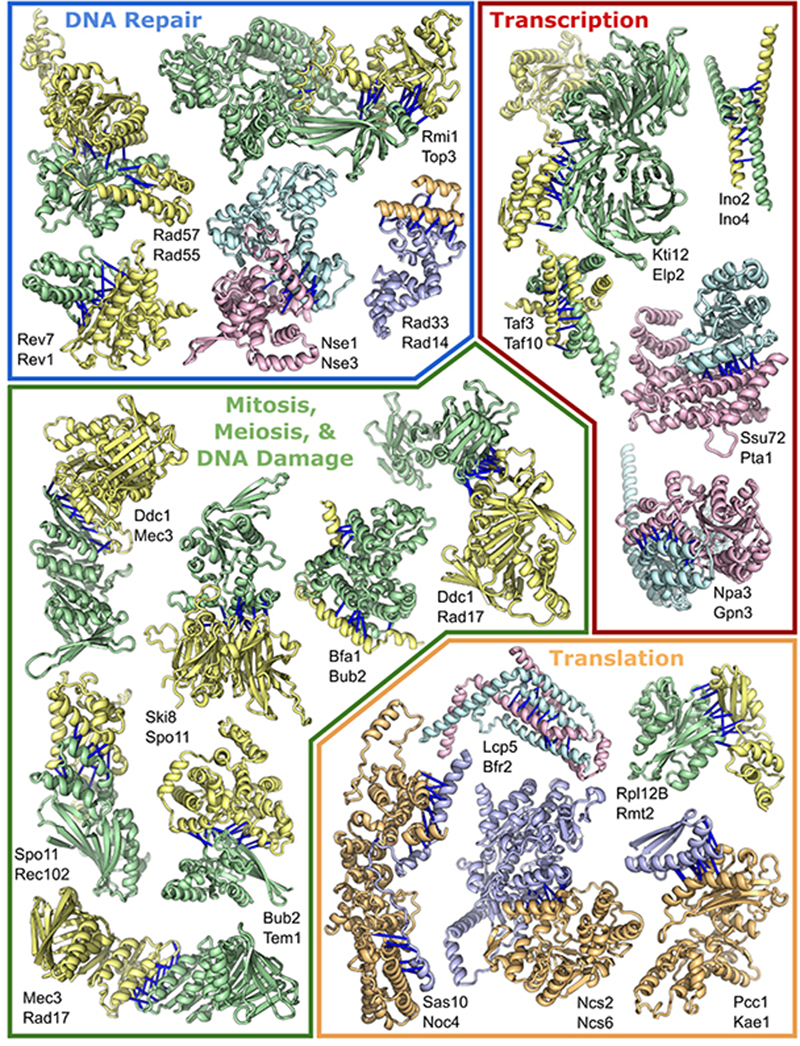
Protein-protein interactions play critical roles in biology, but the structures of many eukaryotic protein complexes are unknown, and there are likely many interactions not yet identified. We take advantage of advances in proteome-wide amino acid coevolution analysis and deep-learning–based structure modeling to systematically identify and build accurate models of core eukaryotic protein complexes within the Saccharomyces cerevisiae proteome. We use a combination of RoseTTAFold and AlphaFold to screen through paired multiple sequence alignments for 8.3 million pairs of yeast proteins, identify 1505 likely to interact, and build structure models for 106 previously unidentified assemblies and 806 that have not been structurally characterized. These complexes, which have as many as five subunits, play roles in almost all key processes in eukaryotic cells and provide broad insights into biological function.
Conflict of interest statement
Figures




Comment in
-
Interactomes in the era of deep learning.Science. 2021 Dec 10;374(6573):1319-1320. doi: 10.1126/science.abm8295. Epub 2021 Dec 9. Science. 2021. PMID: 34882469
References
-
- Collins SR, Kemmeren P, Zhao X-C, Greenblatt JF, Spencer F, Holstege FCP, Weissman JS, Krogan NJ. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol Cell Proteomics. 2007;6:439–450. - PubMed
-
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
