

Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing
- PMID: 34763716
- PMCID: PMC8582192
- DOI: 10.1186/s13059-021-02525-6
Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing
Abstract
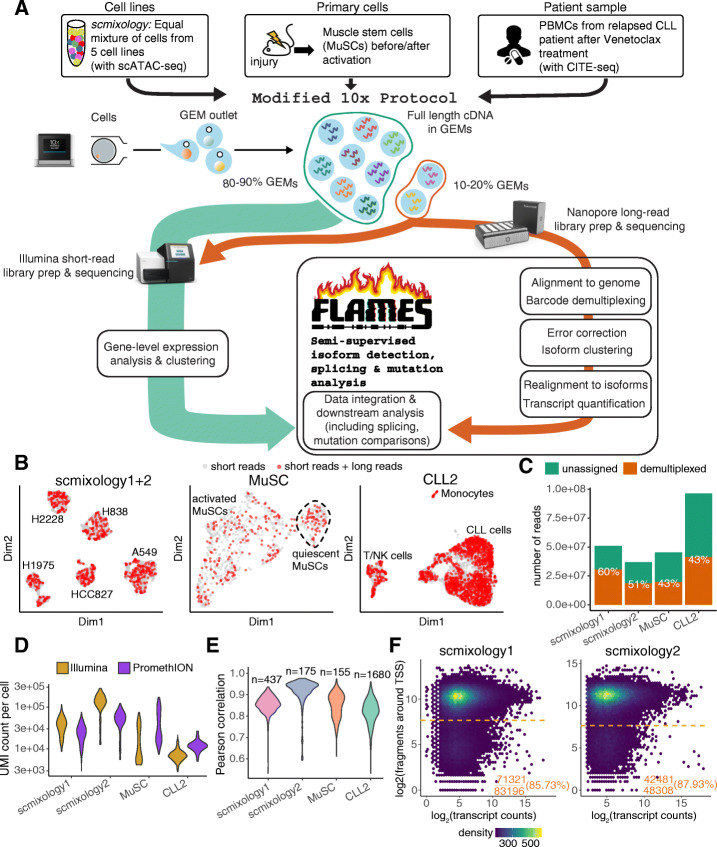
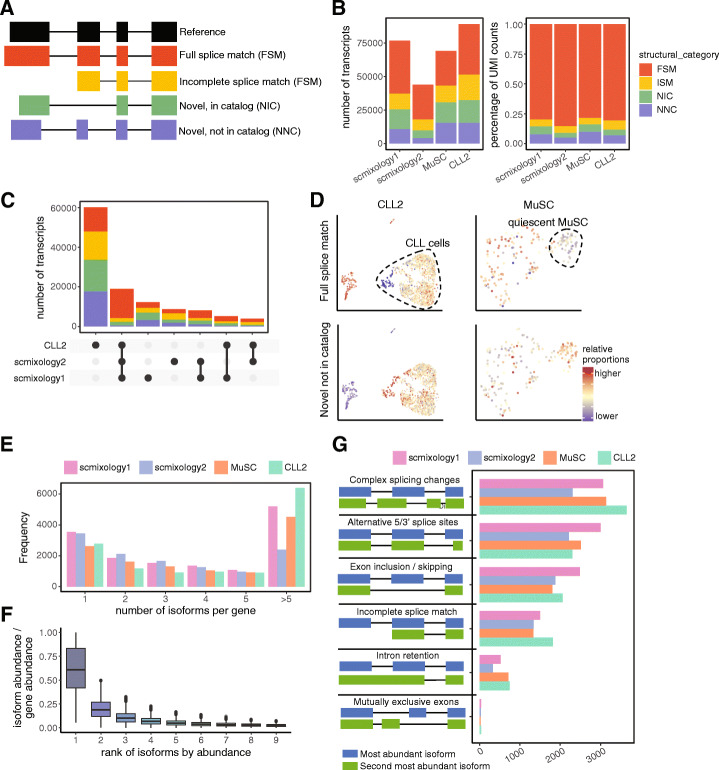
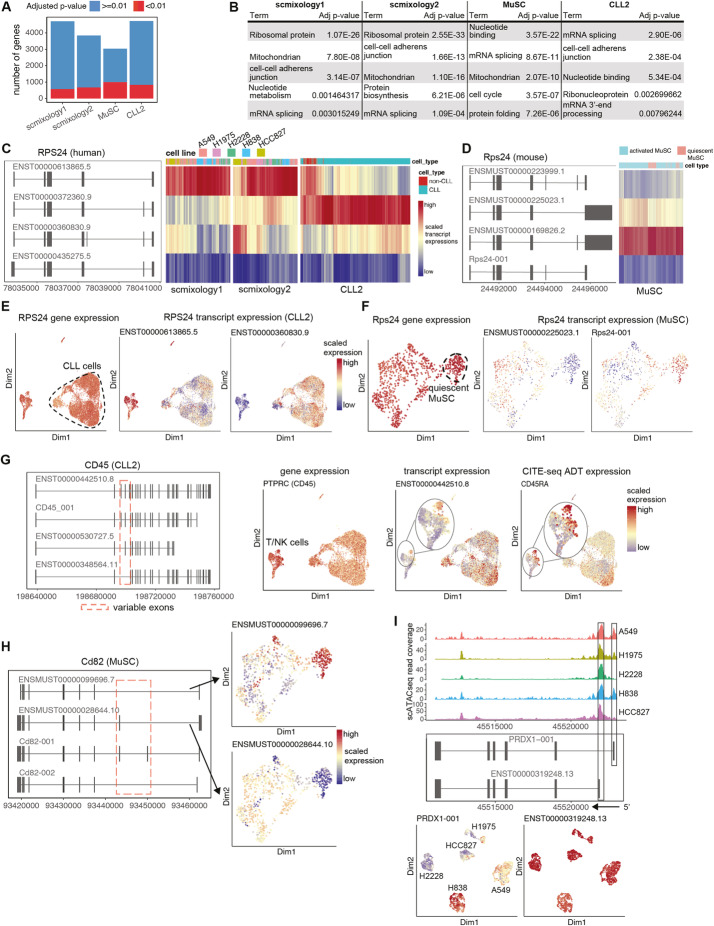
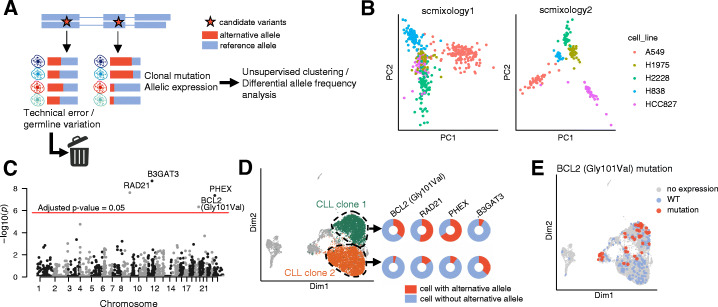
A modified Chromium 10x droplet-based protocol that subsamples cells for both short-read and long-read (nanopore) sequencing together with a new computational pipeline (FLAMES) is developed to enable isoform discovery, splicing analysis, and mutation detection in single cells. We identify thousands of unannotated isoforms and find conserved functional modules that are enriched for alternative transcript usage in different cell types and species, including ribosome biogenesis and mRNA splicing. Analysis at the transcript level allows data integration with scATAC-seq on individual promoters, improved correlation with protein expression data, and linked mutations known to confer drug resistance to transcriptome heterogeneity.
Keywords: Long-read sequencing; Single-cell gene expression; Single-cell multi-omics; Splicing.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y, et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581(7808):303–9. Available from: 10.1038/s41586-020-2157-4. - PubMed
-
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–40. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3683364&tool=p.... - PMC - PubMed
-
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. Available from: http://www.nature.com/articles/ncomms14049. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
