

Long-read assembly and comparative evidence-based reanalysis of Cryptosporidium genome sequences reveal expanded transporter repertoire and duplication of entire chromosome ends including subtelomeric regions
- PMID: 34764149
- PMCID: PMC8744675
- DOI: 10.1101/gr.275325.121
Long-read assembly and comparative evidence-based reanalysis of Cryptosporidium genome sequences reveal expanded transporter repertoire and duplication of entire chromosome ends including subtelomeric regions
Abstract
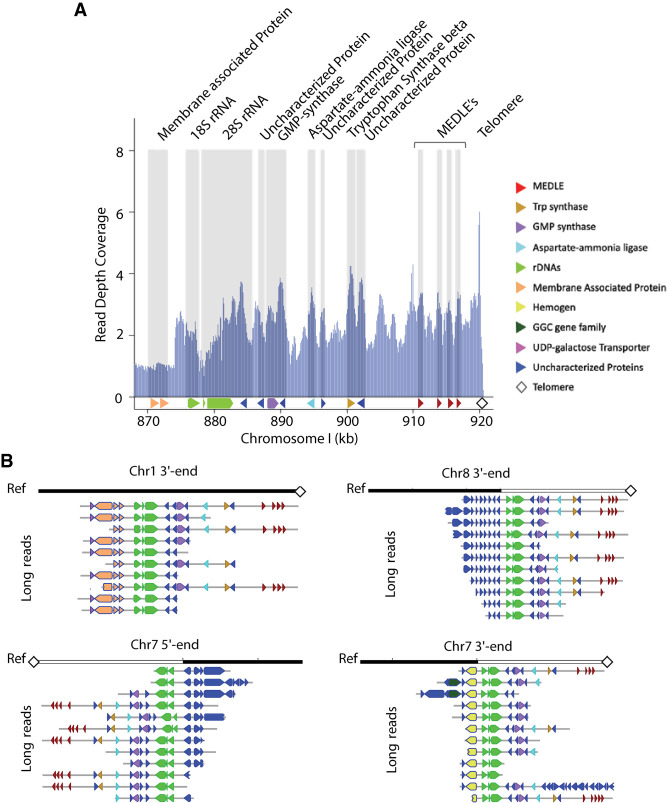
Cryptosporidiosis is a leading cause of waterborne diarrheal disease globally and an important contributor to mortality in infants and the immunosuppressed. Despite its importance, the Cryptosporidium community has only had access to a good, but incomplete, Cryptosporidium parvum IOWA reference genome sequence. Incomplete reference sequences hamper annotation, experimental design, and interpretation. We have generated a new C. parvum IOWA genome assembly supported by Pacific Biosciences (PacBio) and Oxford Nanopore long-read technologies and a new comparative and consistent genome annotation for three closely related species: C. parvum, Cryptosporidium hominis, and Cryptosporidium tyzzeri We made 1926 C. parvum annotation updates based on experimental evidence. They include new transporters, ncRNAs, introns, and altered gene structures. The new assembly and annotation revealed a complete Dnmt2 methylase ortholog. Comparative annotation between C. parvum, C. hominis, and C. tyzzeri revealed that most "missing" orthologs are found, suggesting that the biological differences between the species must result from gene copy number variation, differences in gene regulation, and single-nucleotide variants (SNVs). Using the new assembly and annotation as reference, 190 genes are identified as evolving under positive selection, including many not detected previously. The new C. parvum IOWA reference genome assembly is larger, gap free, and lacks ambiguous bases. This chromosomal assembly recovers all 16 chromosome ends, 13 of which are contiguously assembled. The three remaining chromosome ends are provisionally placed. These ends represent duplication of entire chromosome ends including subtelomeric regions revealing a new level of genome plasticity that will both inform and impact future research.
© 2022 Baptista et al.; Published by Cold Spring Harbor Laboratory Press.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous