

Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances
- PMID: 34768808
- PMCID: PMC8583656
- DOI: 10.3390/ijms222111377
Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances
Abstract
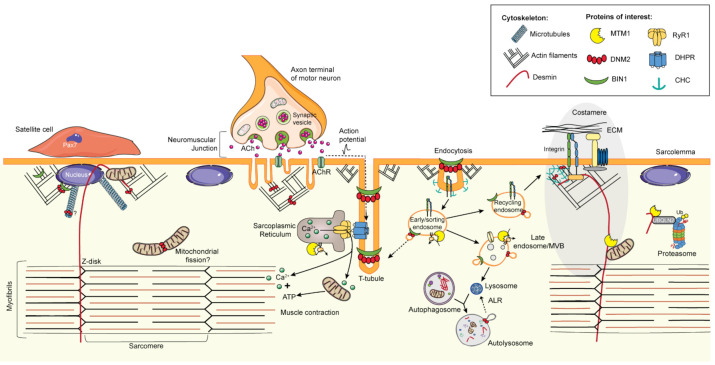
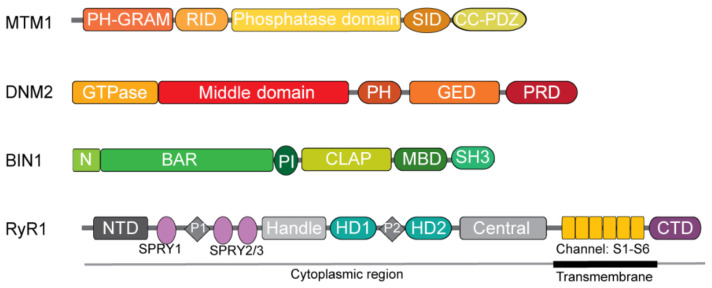
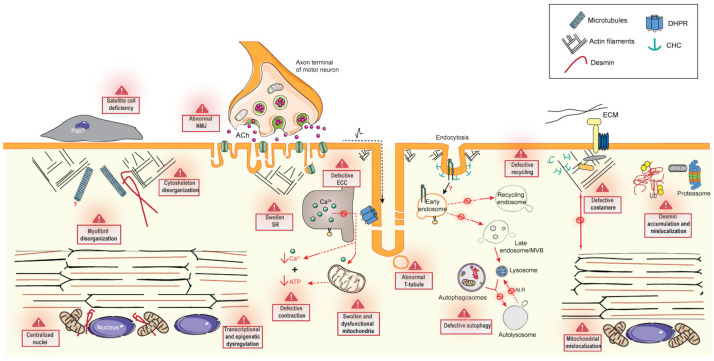
Centronuclear myopathies (CNM) are rare congenital disorders characterized by muscle weakness and structural defects including fiber hypotrophy and organelle mispositioning. The main CNM forms are caused by mutations in: the MTM1 gene encoding the phosphoinositide phosphatase myotubularin (myotubular myopathy), the DNM2 gene encoding the mechanoenzyme dynamin 2, the BIN1 gene encoding the membrane curvature sensing amphiphysin 2, and the RYR1 gene encoding the skeletal muscle calcium release channel/ryanodine receptor. MTM1, BIN1, and DNM2 proteins are involved in membrane remodeling and trafficking, while RyR1 directly regulates excitation-contraction coupling (ECC). Several CNM animal models have been generated or identified, which confirm shared pathological anomalies in T-tubule remodeling, ECC, organelle mispositioning, protein homeostasis, neuromuscular junction, and muscle regeneration. Dynamin 2 plays a crucial role in CNM physiopathology and has been validated as a common therapeutic target for three CNM forms. Indeed, the promising results in preclinical models set up the basis for ongoing clinical trials. Another two clinical trials to treat myotubular myopathy by MTM1 gene therapy or tamoxifen repurposing are also ongoing. Here, we review the contribution of the different CNM models to understanding physiopathology and therapy development with a focus on the commonly dysregulated pathways and current therapeutic targets.
Keywords: amphiphysin; autophagy; centronuclear myopathy; dynamin; membrane trafficking; myotubular myopathy; myotubularin; ryanodine receptor; satellite cell; triads.
Conflict of interest statement
B.S. Cowling and J. Laporte are co-founders of Dynacure. R. Gomez-Oca and B.S. Cowling are employees of Dynacure.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
