

Deacetylation of Transcription Factors in Carcinogenesis
- PMID: 34769241
- PMCID: PMC8583941
- DOI: 10.3390/ijms222111810
Deacetylation of Transcription Factors in Carcinogenesis
Abstract
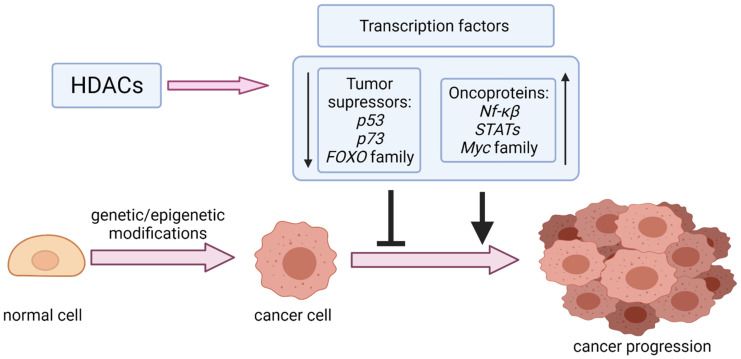
Reversible Nε-lysine acetylation/deacetylation is one of the most common post-translational modifications (PTM) of histones and non-histone proteins that is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). This epigenetic process is highly involved in carcinogenesis, affecting histone and non-histone proteins' properties and their biological functions. Some of the transcription factors, including tumor suppressors and oncoproteins, undergo this modification altering different cell signaling pathways. HDACs deacetylate their targets, which leads to either the upregulation or downregulation of proteins involved in the regulation of cell cycle and apoptosis, ultimately influencing tumor growth, invasion, and drug resistance. Therefore, epigenetic modifications are of great clinical importance and may constitute a new therapeutic target in cancer treatment. This review is aimed to present the significance of HDACs in carcinogenesis through their influence on functions of transcription factors, and therefore regulation of different signaling pathways, cancer progression, and metastasis.
Keywords: HDAC; histone deacetylase inhibitors (HDIs); transcription factors.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- (US) NI of H, Study BSC Understanding Cancer. [(accessed on 3 September 2021)];2007 Available online: https://www.ncbi.nlm.nih.gov/books/NBK20362/
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
