

In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies
- PMID: 34771004
- PMCID: PMC8588135
- DOI: 10.3390/molecules26216593
In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies
Abstract
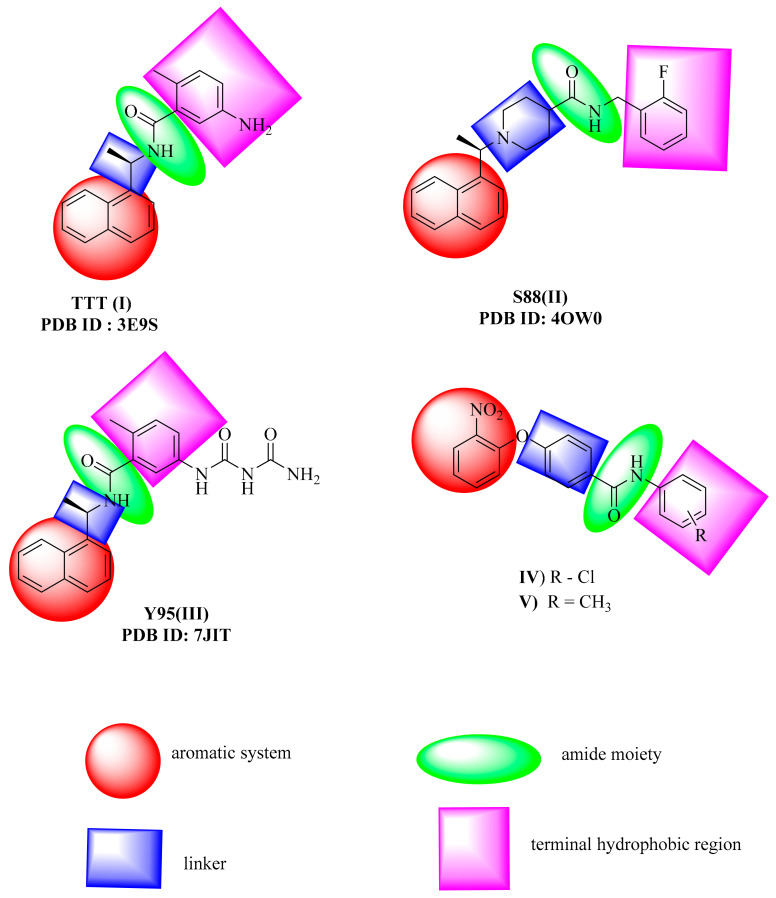
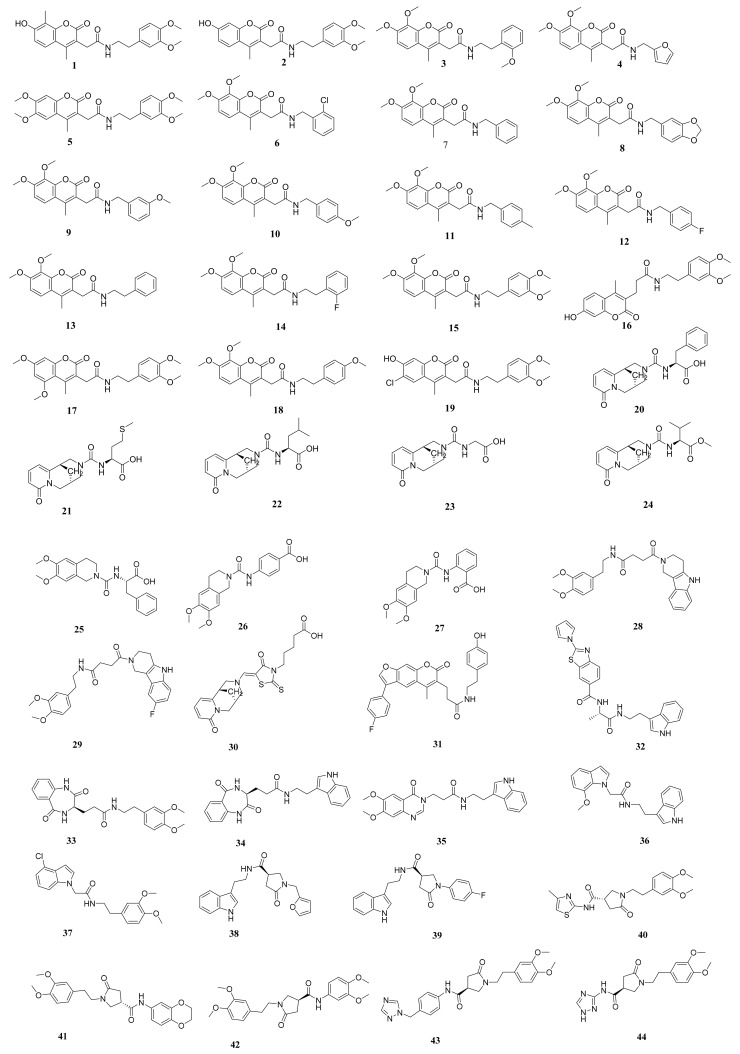
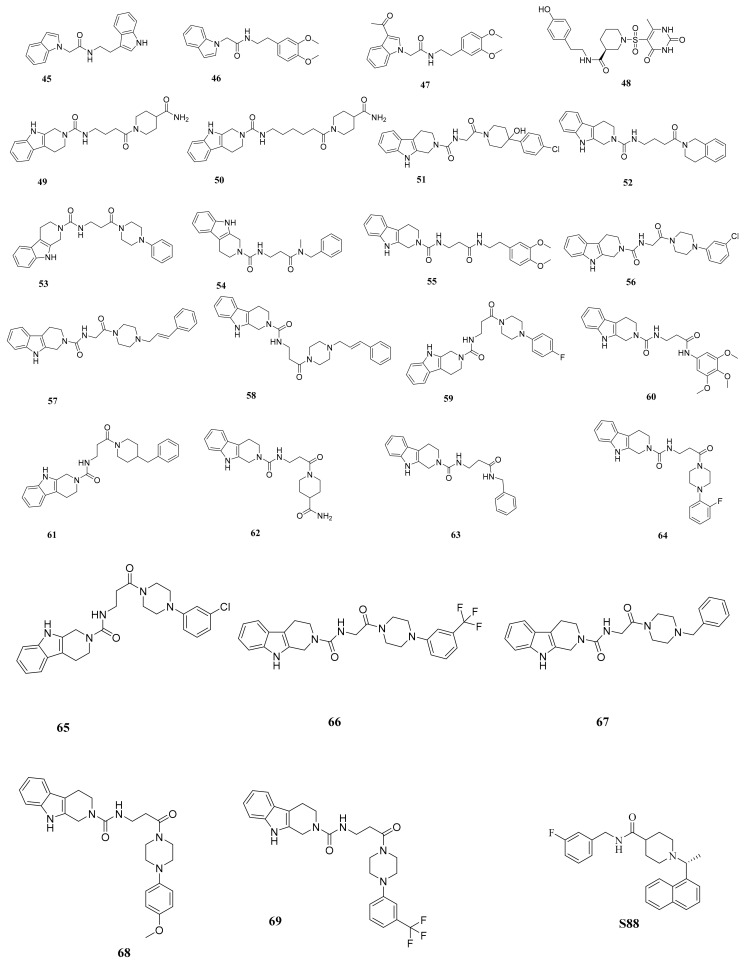
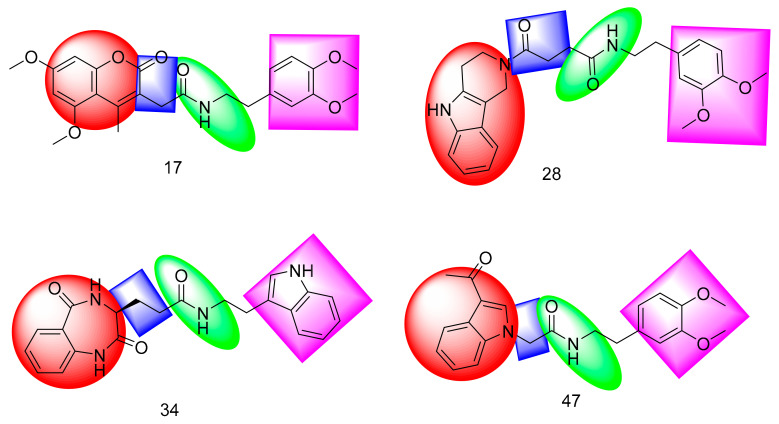
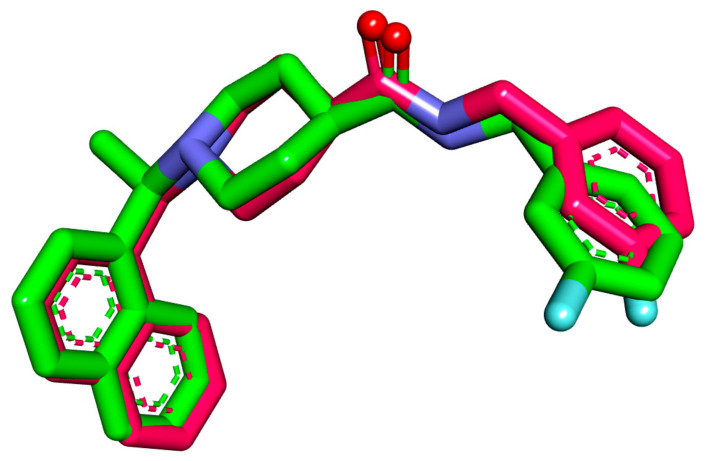
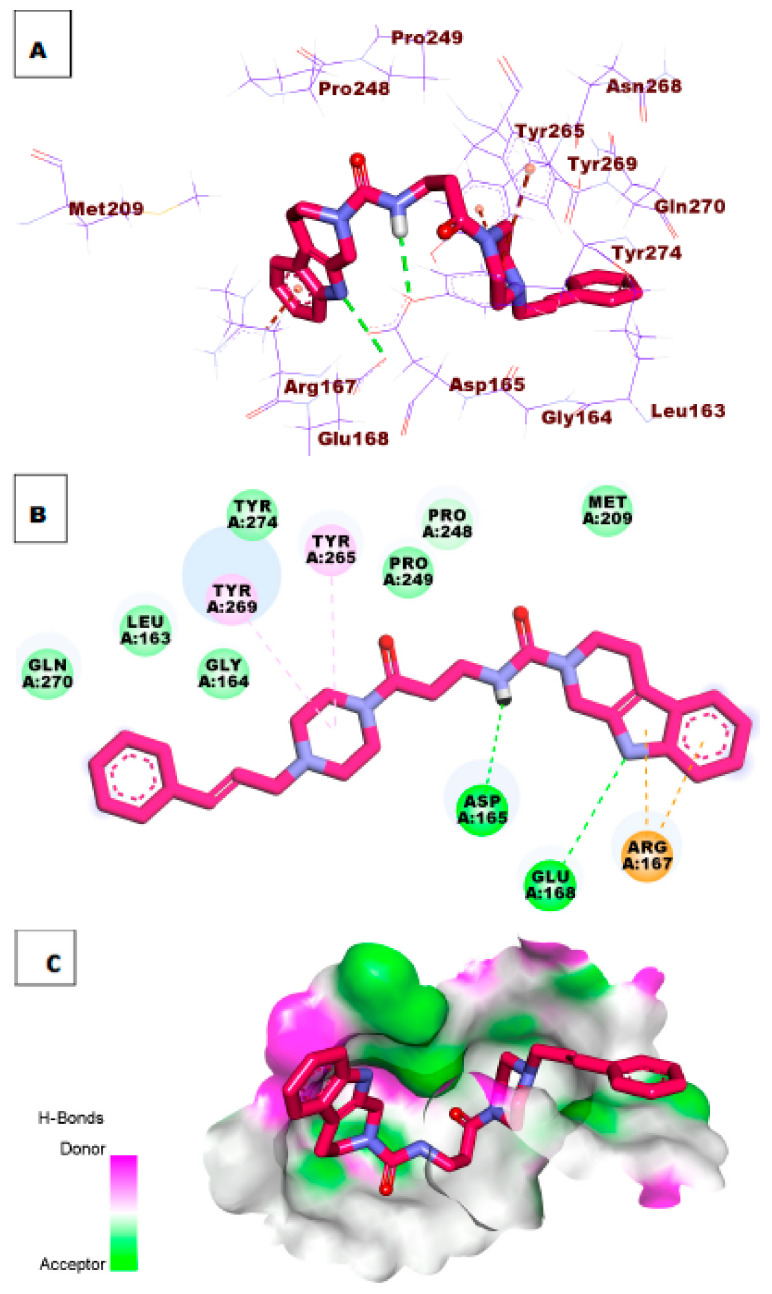
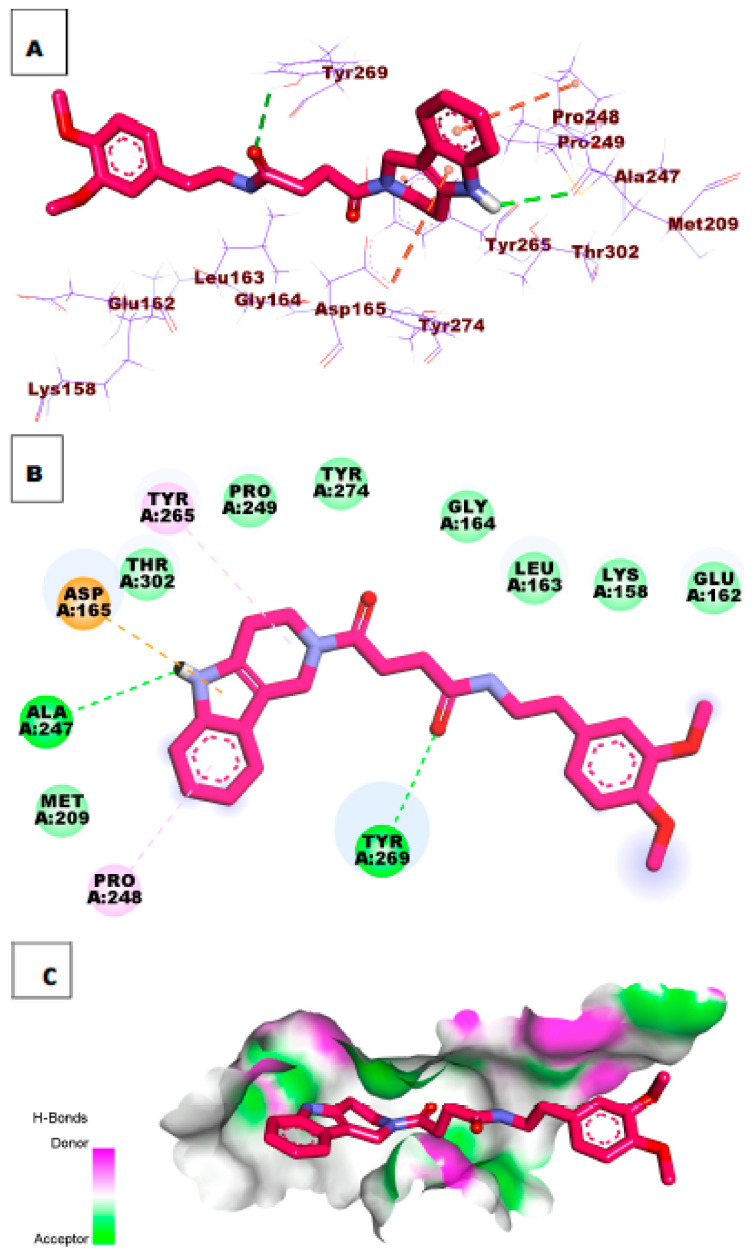
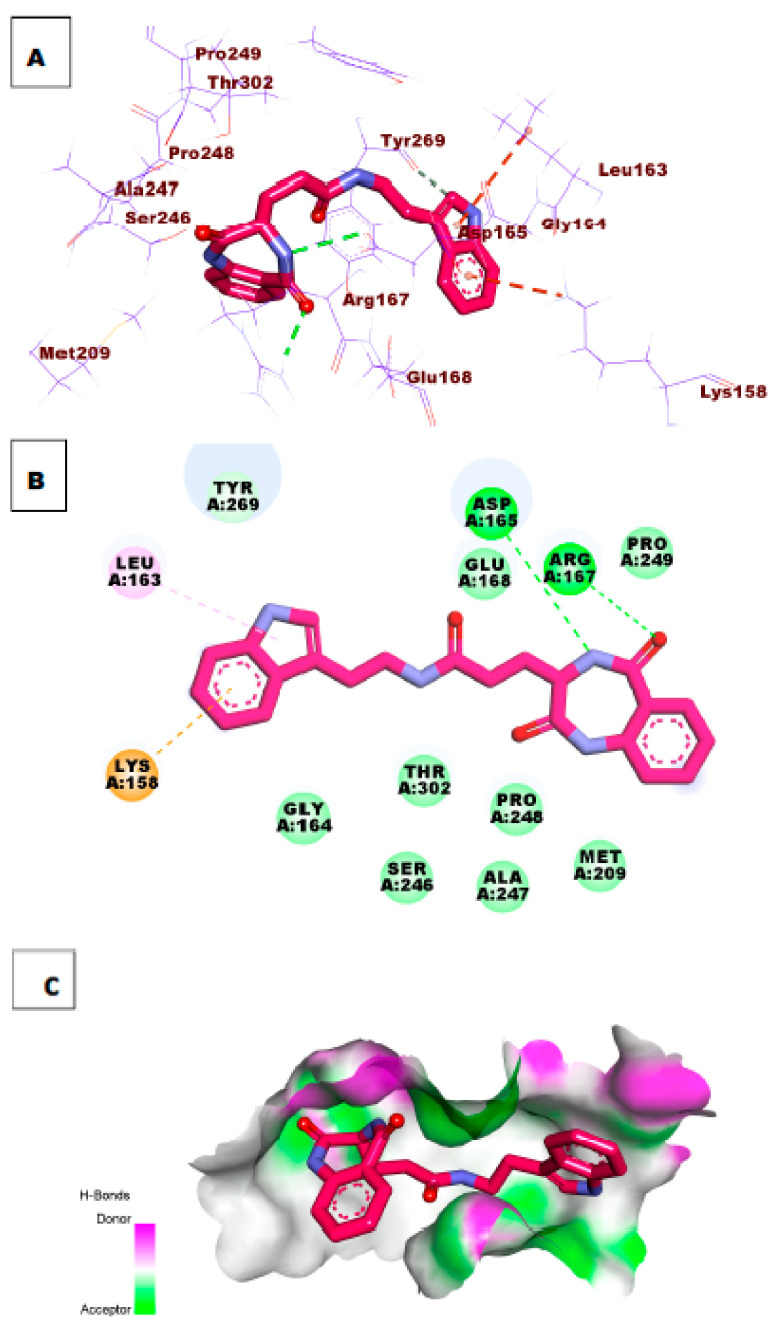
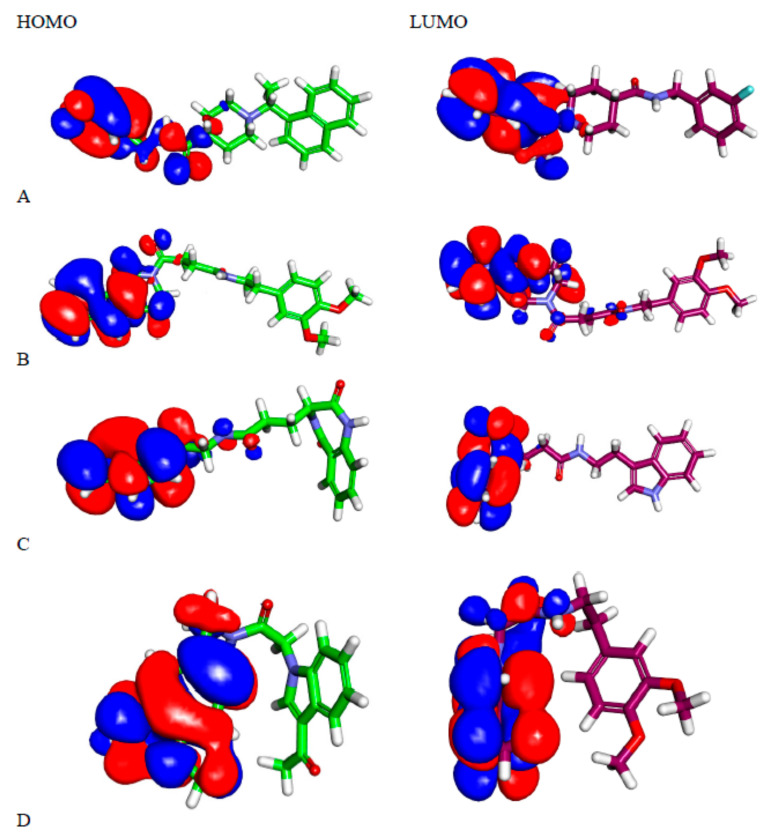
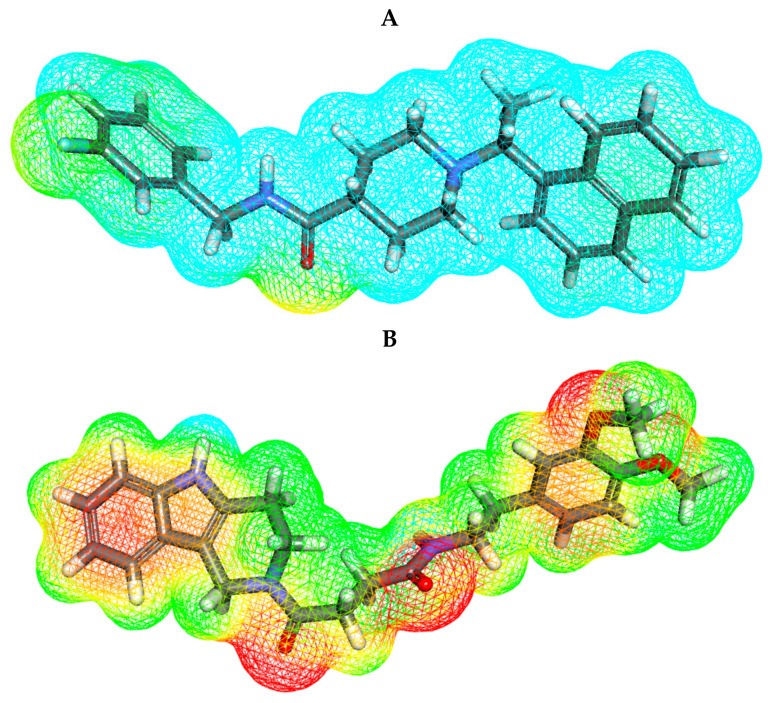
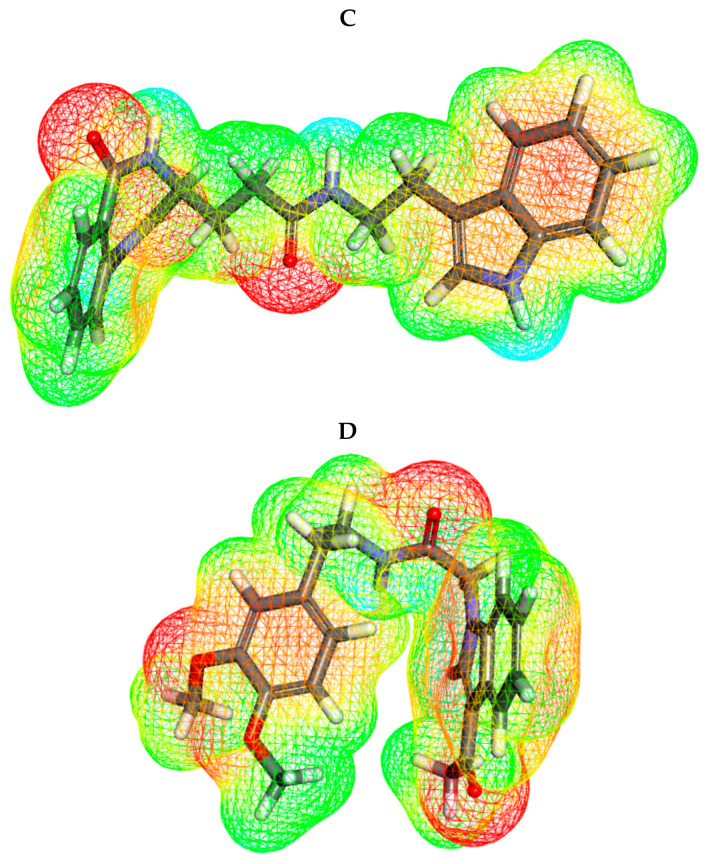
Papain-like protease is an essential enzyme in the proteolytic processing required for the replication of SARS-CoV-2. Accordingly, such an enzyme is an important target for the development of anti-SARS-CoV-2 agents which may reduce the mortality associated with outbreaks of SARS-CoV-2. A set of 69 semi-synthesized molecules that exhibited the structural features of SARS-CoV-2 papain-like protease inhibitors (PLPI) were docked against the coronavirus papain-like protease (PLpro) enzyme (PDB ID: (4OW0). Docking studies showed that derivatives 34 and 58 were better than the co-crystallized ligand while derivatives 17, 28, 31, 40, 41, 43, 47, 54, and 65 exhibited good binding modes and binding free energies. The pharmacokinetic profiling study was conducted according to the four principles of the Lipinski rules and excluded derivative 31. Furthermore, ADMET and toxicity studies showed that derivatives 28, 34, and 47 have the potential to be drugs and have been demonstrated as safe when assessed via seven toxicity models. Finally, comparing the molecular orbital energies and the molecular electrostatic potential maps of 28, 34, and 47 against the co-crystallized ligand in a DFT study indicated that 28 is the most promising candidate to interact with the target receptor (PLpro).
Keywords: ADMET; COVID-19; DFT; molecular docking; papain-like protease; pharmacophore; semi-synthesized; toxicity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
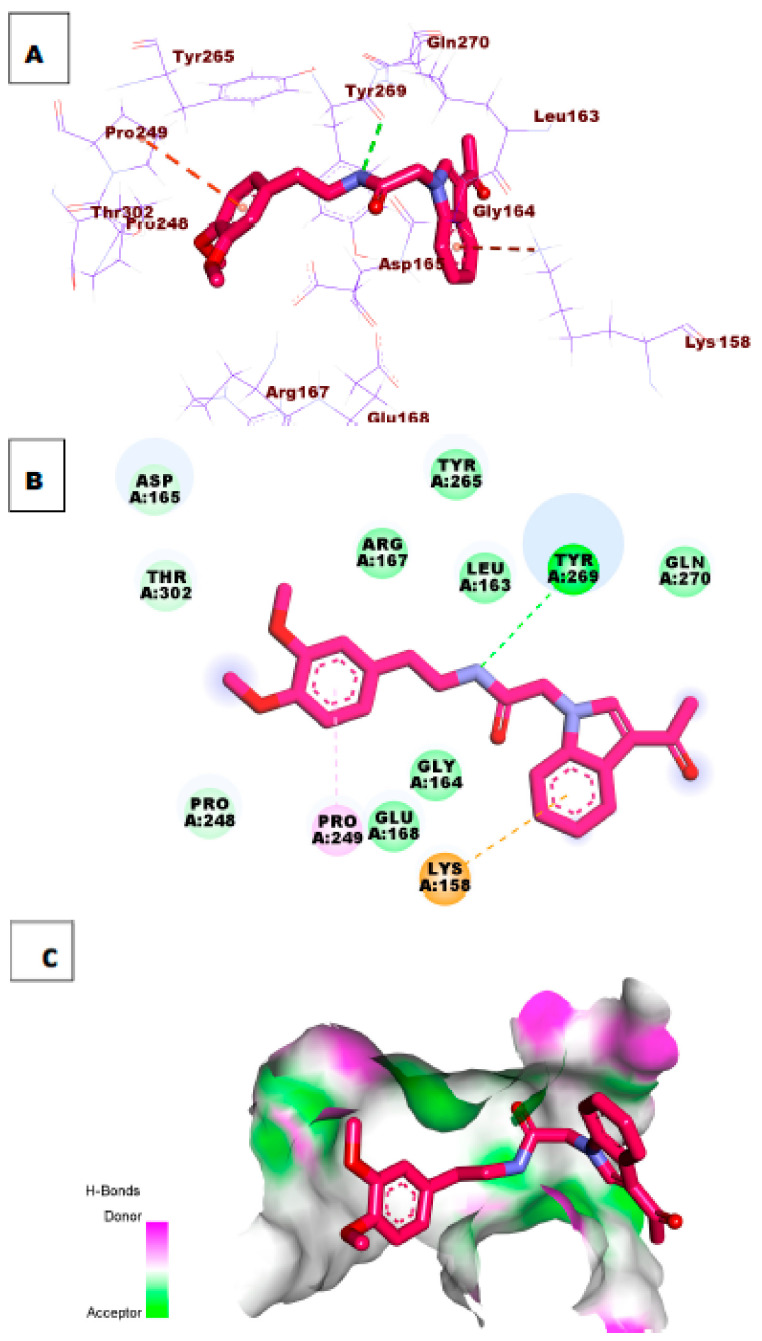
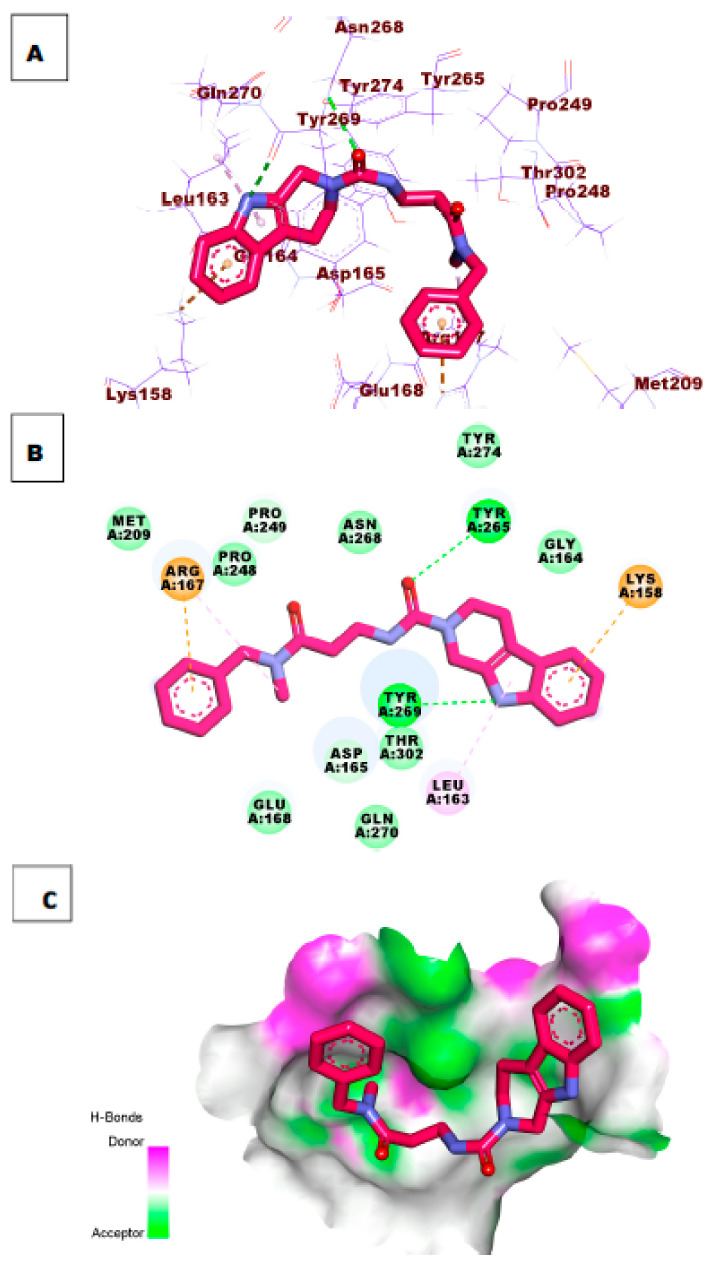
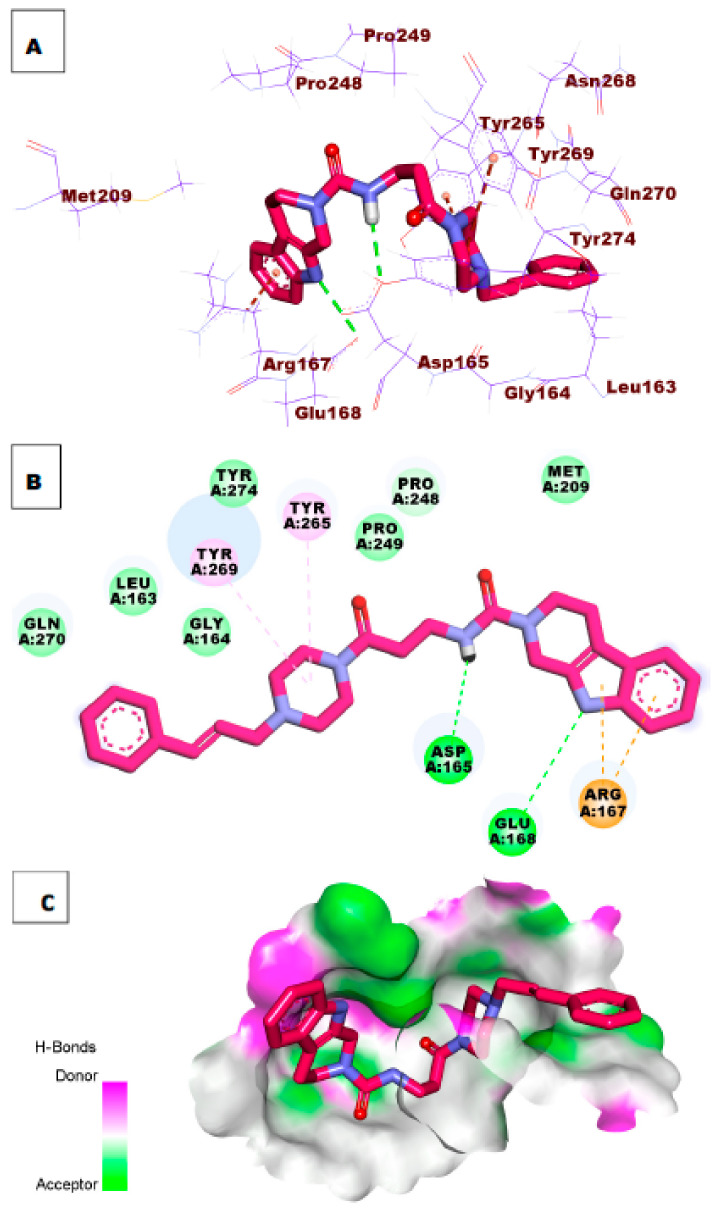
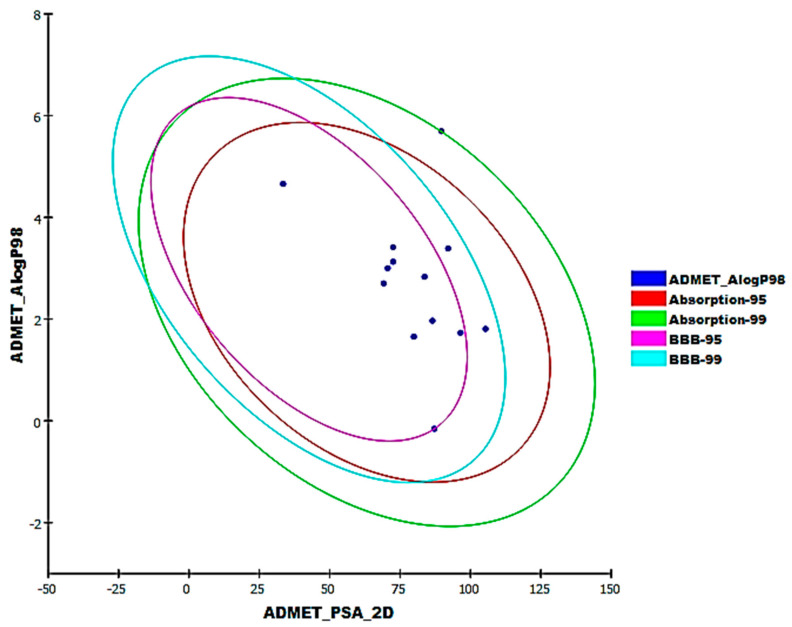
References
-
- WHO WHO Coronavirus (COVID-19) Dashboard. [(accessed on 10 September 2021)]. Available online: https://covid19.who.int/
-
- Li N., Wang Y., Li W., Li H., Yang L., Wang J., Mahdy H.A., Mehany A., Jaiash D.A., Santali E.Y. Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem. 2020;2020:1631243. doi: 10.1155/2020/1631243. - DOI
-
- Abdel-Aziz H.A., Eldehna W.M., Fares M., Al-Rashood S.T., Al-Rashood K.A., Abdel-Aziz M.M., Soliman D.H. Synthesis, biological evaluation and 2D-QSAR study of halophenyl bis-hydrazones as antimicrobial and antitubercular agents. Int. J. Mol. Sci. 2015;16:8719–8743. doi: 10.3390/ijms16048719. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
