

EP300 Selectively Controls the Enhancer Landscape of MYCN-Amplified Neuroblastoma
- PMID: 34772733
- PMCID: PMC8904277
- DOI: 10.1158/2159-8290.CD-21-0385
EP300 Selectively Controls the Enhancer Landscape of MYCN-Amplified Neuroblastoma
Abstract
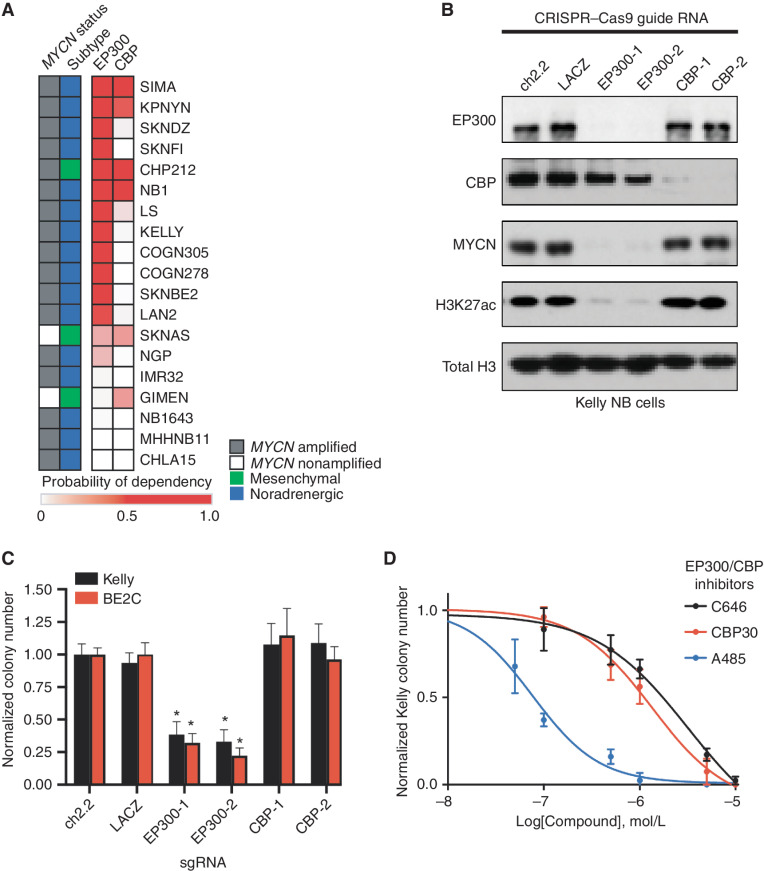
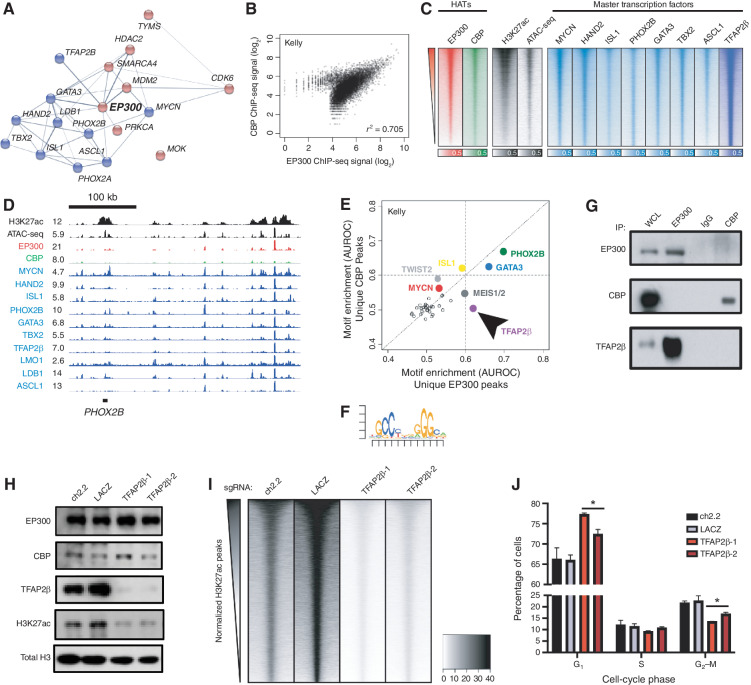
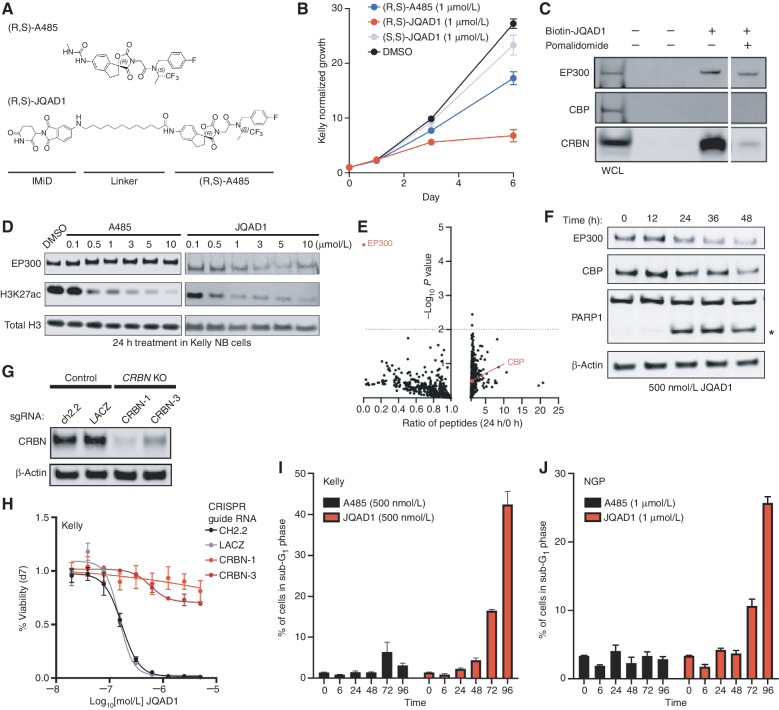
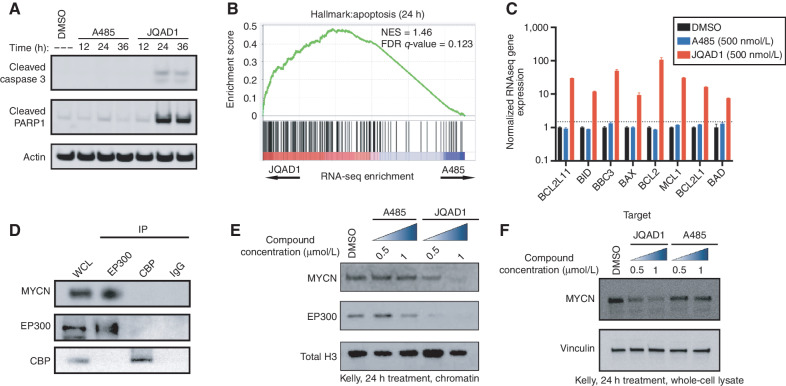
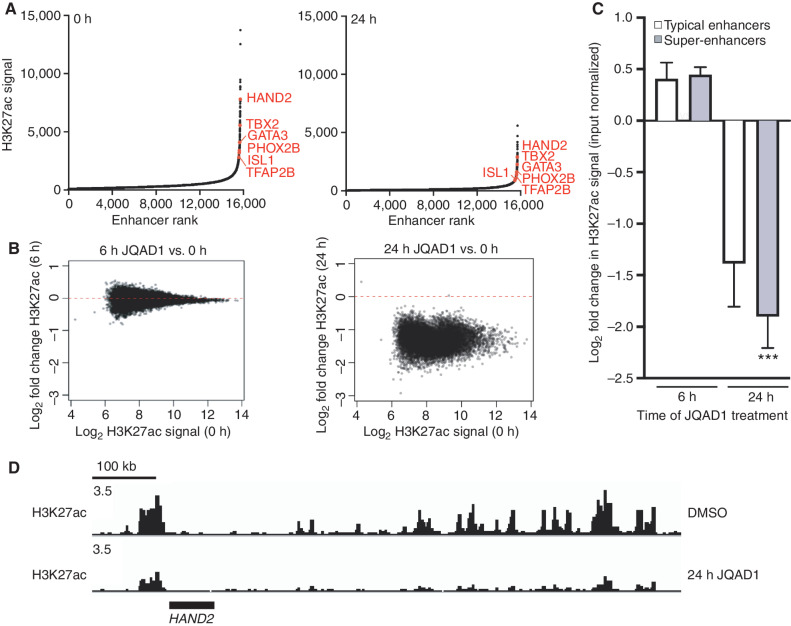
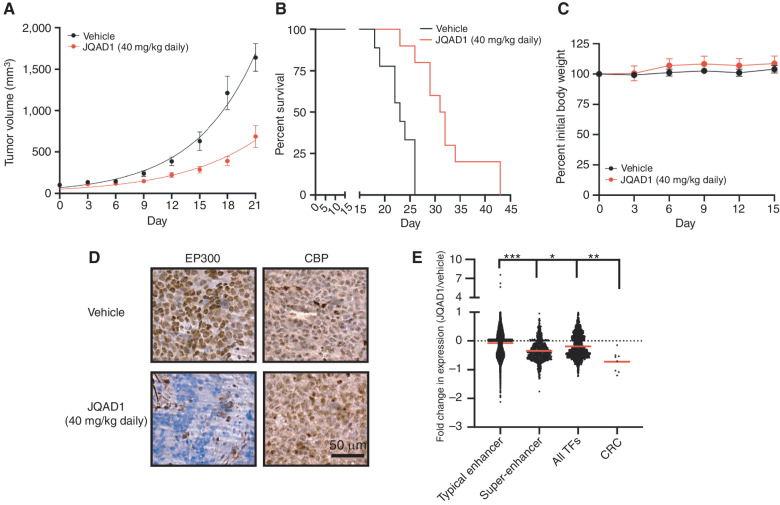
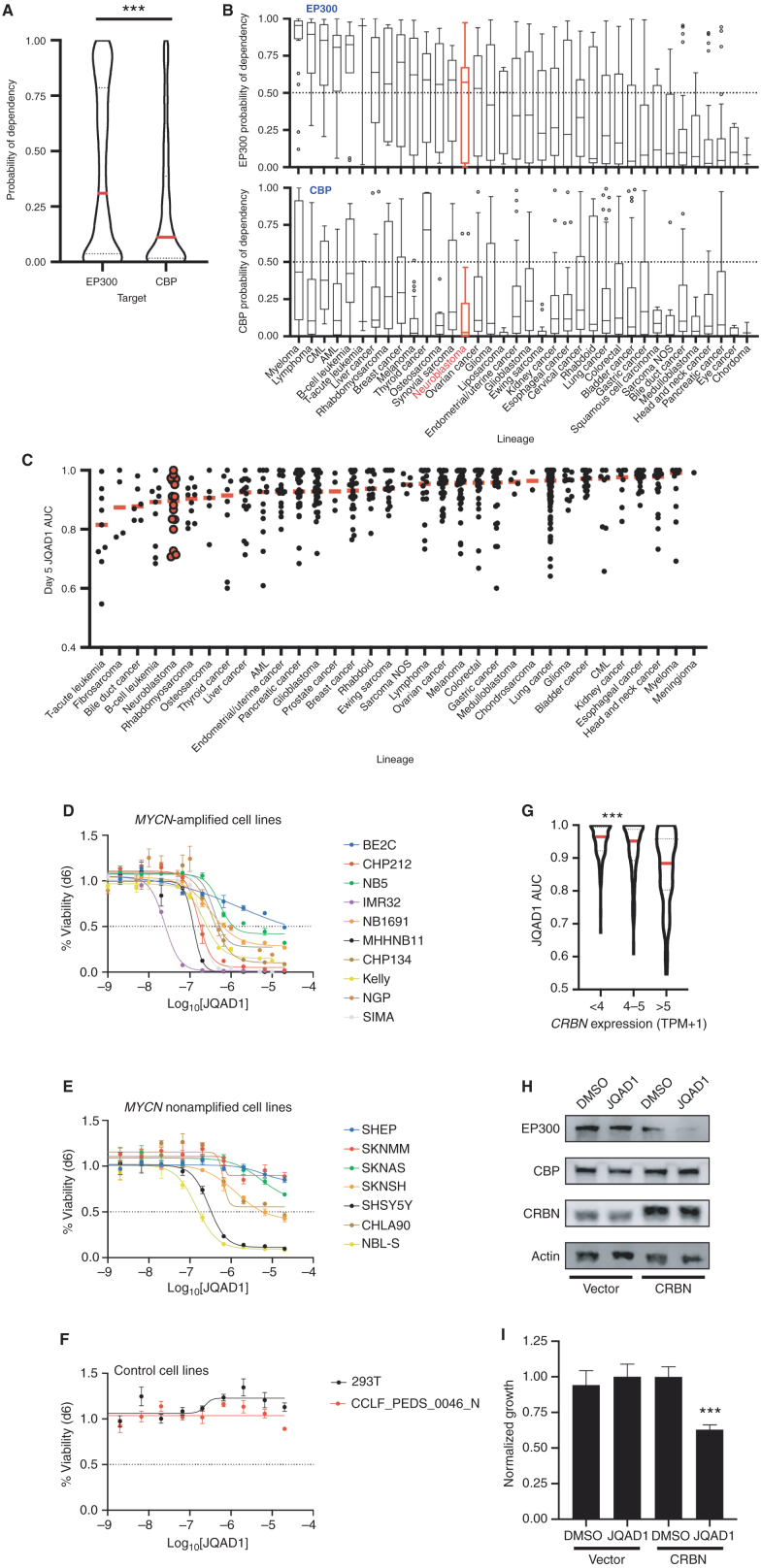
Gene expression is regulated by promoters and enhancers marked by histone H3 lysine 27 acetylation (H3K27ac), which is established by the paralogous histone acetyltransferases (HAT) EP300 and CBP. These enzymes display overlapping regulatory roles in untransformed cells, but less characterized roles in cancer cells. We demonstrate that the majority of high-risk pediatric neuroblastoma (NB) depends on EP300, whereas CBP has a limited role. EP300 controls enhancer acetylation by interacting with TFAP2β, a transcription factor member of the lineage-defining transcriptional core regulatory circuitry (CRC) in NB. To disrupt EP300, we developed a proteolysis-targeting chimera (PROTAC) compound termed "JQAD1" that selectively targets EP300 for degradation. JQAD1 treatment causes loss of H3K27ac at CRC enhancers and rapid NB apoptosis, with limited toxicity to untransformed cells where CBP may compensate. Furthermore, JQAD1 activity is critically determined by cereblon (CRBN) expression across NB cells.
Significance: EP300, but not CBP, controls oncogenic CRC-driven transcription in high-risk NB by binding TFAP2β. We developed JQAD1, a CRBN-dependent PROTAC degrader with preferential activity against EP300 and demonstrated its activity in NB. JQAD1 has limited toxicity to untransformed cells and is effective in vivo in a CRBN-dependent manner. This article is highlighted in the In This Issue feature, p. 587.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
- Cancer Discov. 12:587.
- Cancer Discov. 12:587.
References
-
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu Met al. The human transcription factors. Cell 2018;175:598–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- T32 CA136432/CA/NCI NIH HHS/United States
- R01 HG010501/HG/NHGRI NIH HHS/United States
- P50 CA206963/CA/NCI NIH HHS/United States
- P01 CA217959/CA/NCI NIH HHS/United States
- P30 CA021765/CA/NCI NIH HHS/United States
- R21 HG009268/HG/NHGRI NIH HHS/United States
- R35 CA210064/CA/NCI NIH HHS/United States
- DRSG-24-18/DRCRF/Damon Runyon Cancer Research Foundation/United States
- R25 CA023944/CA/NCI NIH HHS/United States
- K08 CA245251/CA/NCI NIH HHS/United States
- R01 NS088355/NS/NINDS NIH HHS/United States
- R35 CA210030/CA/NCI NIH HHS/United States
- P01 CA066996/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
