

Local DNA shape is a general principle of transcription factor binding specificity in Arabidopsis thaliana
- PMID: 34772949
- PMCID: PMC8590021
- DOI: 10.1038/s41467-021-26819-2
Local DNA shape is a general principle of transcription factor binding specificity in Arabidopsis thaliana
Abstract
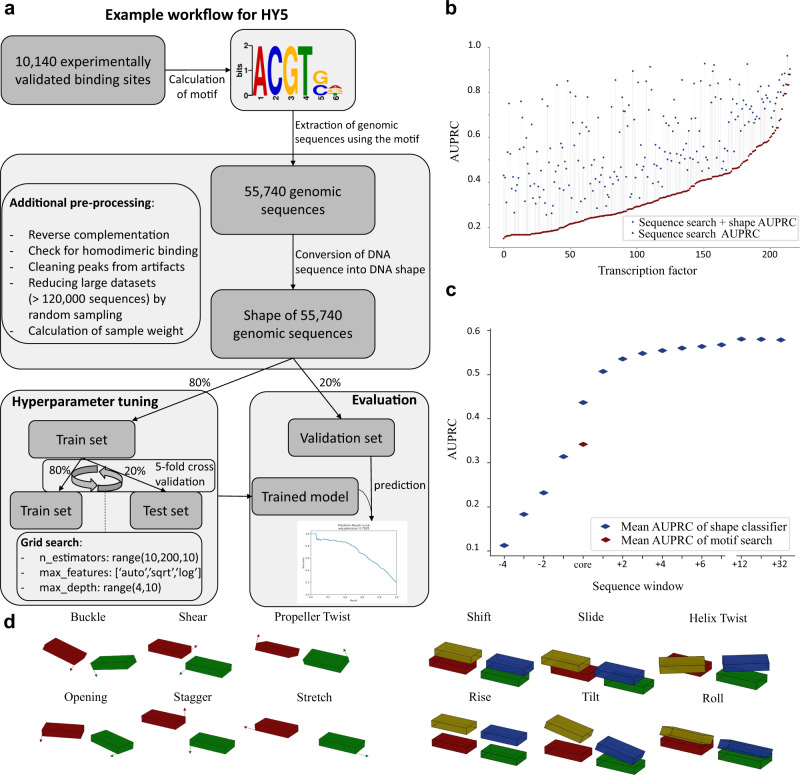
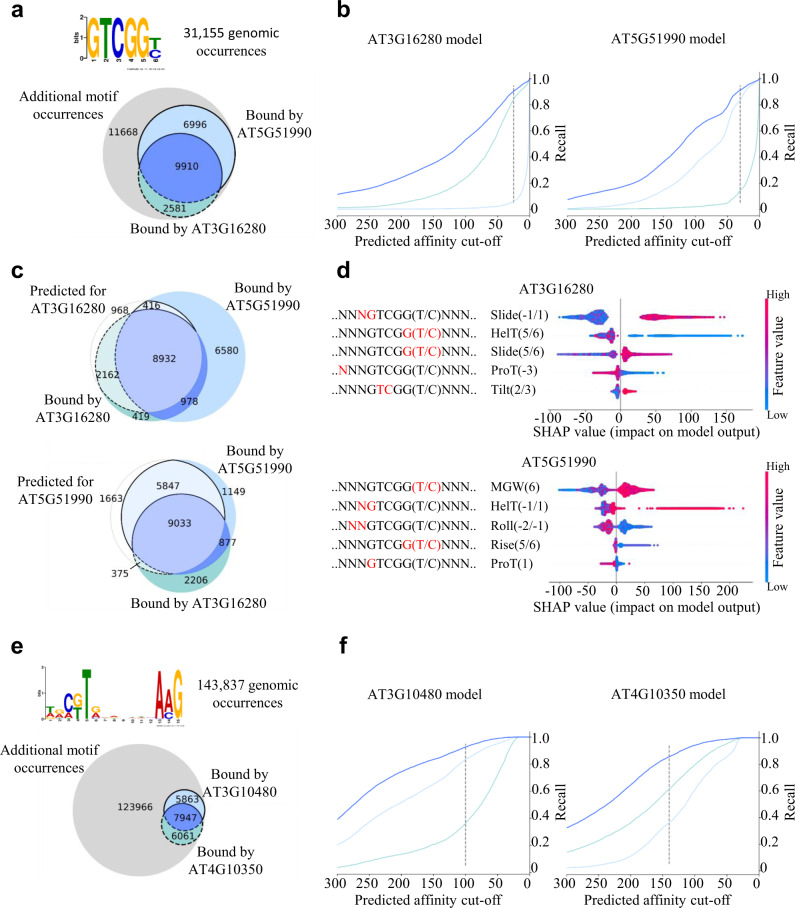
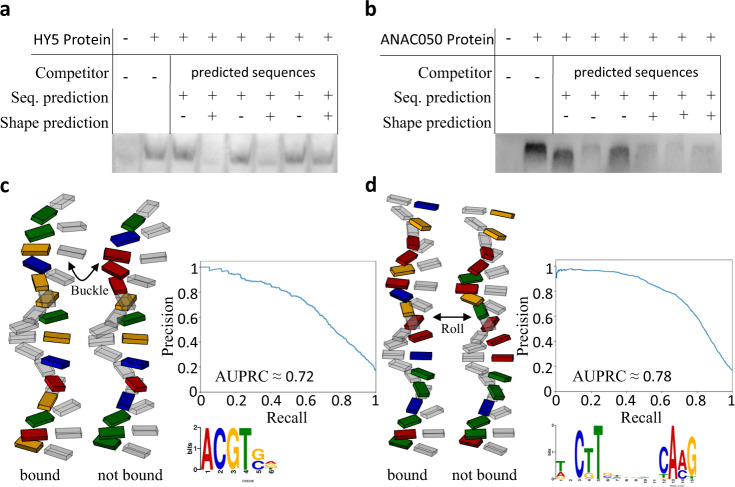
Understanding gene expression will require understanding where regulatory factors bind genomic DNA. The frequently used sequence-based motifs of protein-DNA binding are not predictive, since a genome contains many more binding sites than are actually bound and transcription factors of the same family share similar DNA-binding motifs. Traditionally, these motifs only depict sequence but neglect DNA shape. Since shape may contribute non-linearly and combinational to binding, machine learning approaches ought to be able to better predict transcription factor binding. Here we show that a random forest machine learning approach, which incorporates the 3D-shape of DNA, enhances binding prediction for all 216 tested Arabidopsis thaliana transcription factors and improves the resolution of differential binding by transcription factor family members which share the same binding motif. We observed that DNA shape features were individually weighted for each transcription factor, even if they shared the same binding sequence.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
