

Differences in local anaesthetic and antiepileptic binding in the inactivated state of human sodium channel Nav1.4
- PMID: 34774501
- PMCID: PMC8715241
- DOI: 10.1016/j.bpj.2021.11.014
Differences in local anaesthetic and antiepileptic binding in the inactivated state of human sodium channel Nav1.4
Abstract
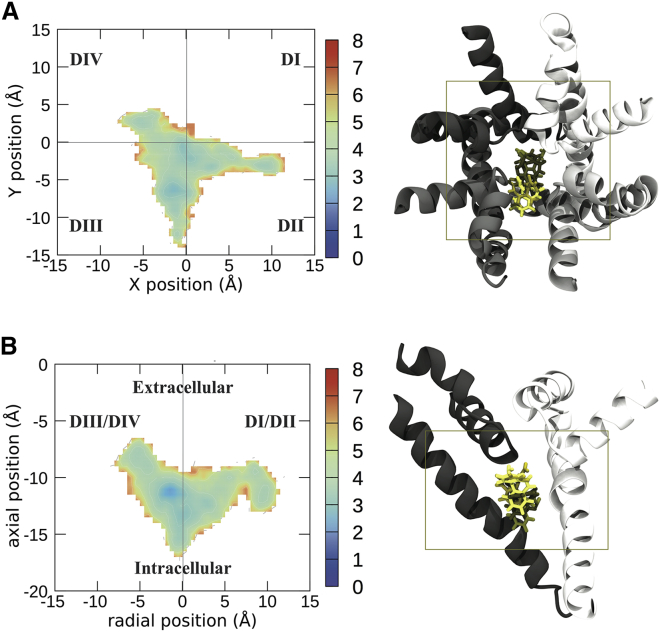
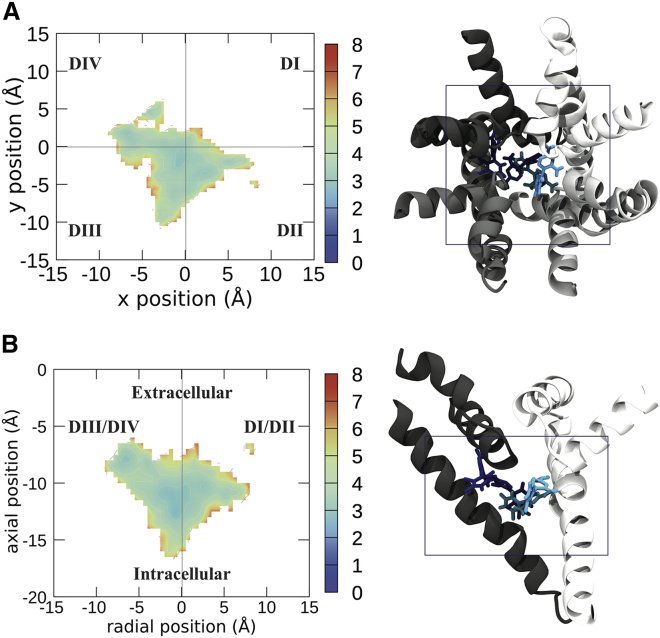
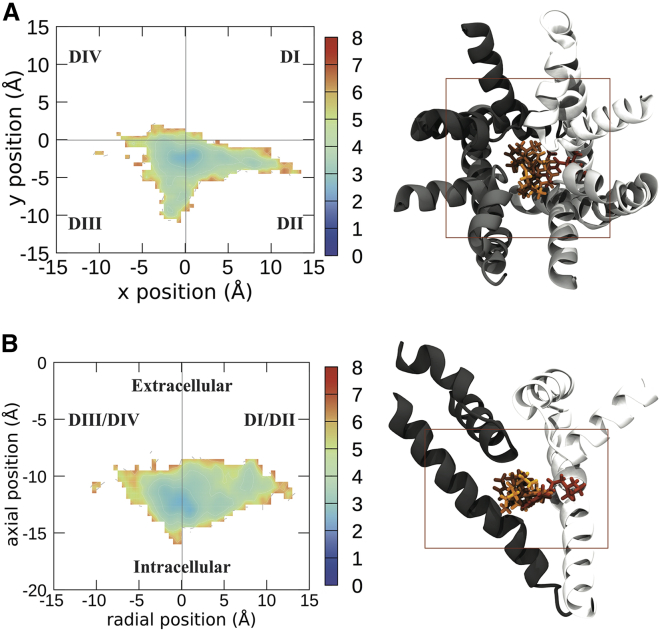
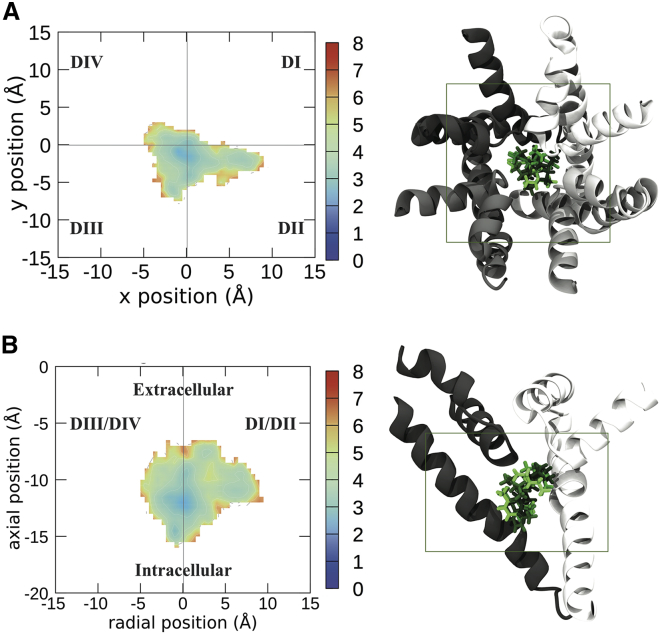
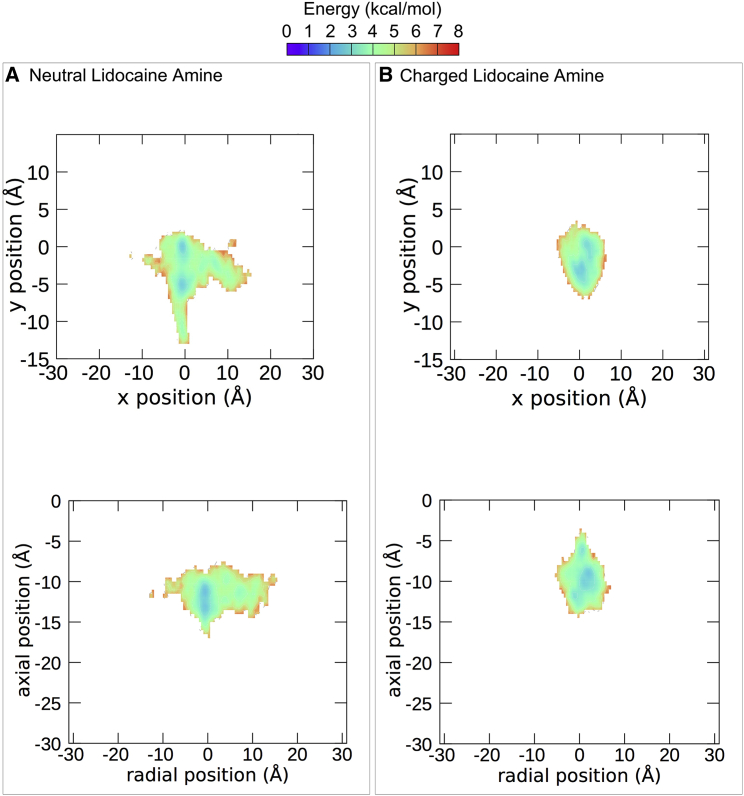
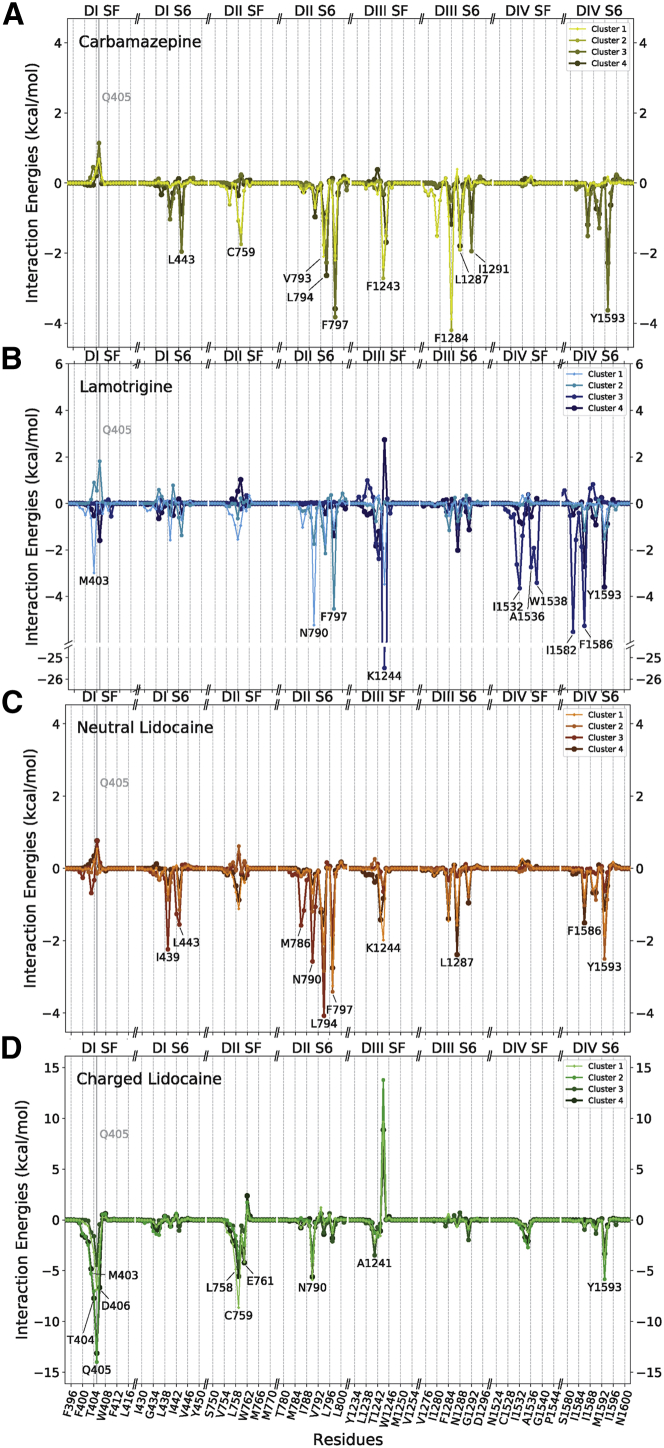
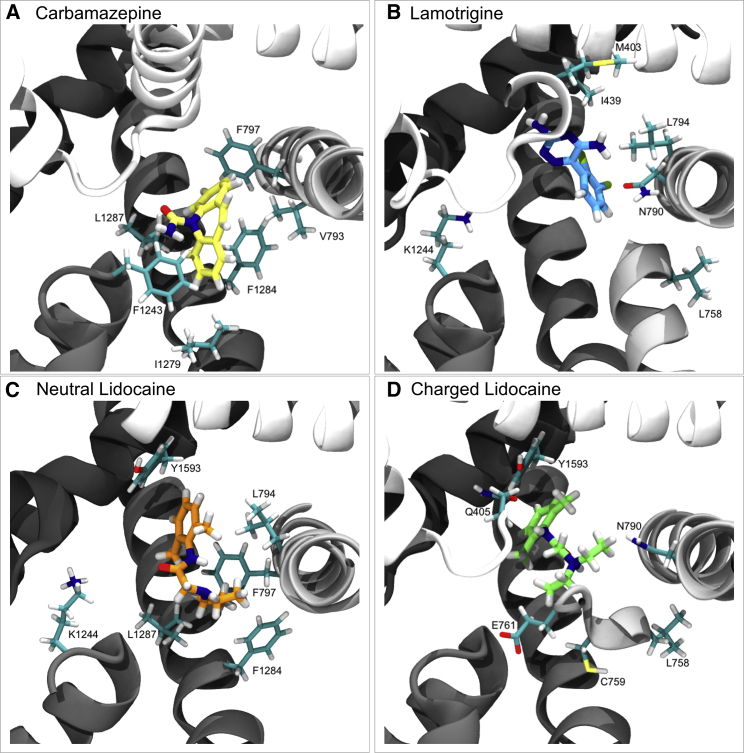
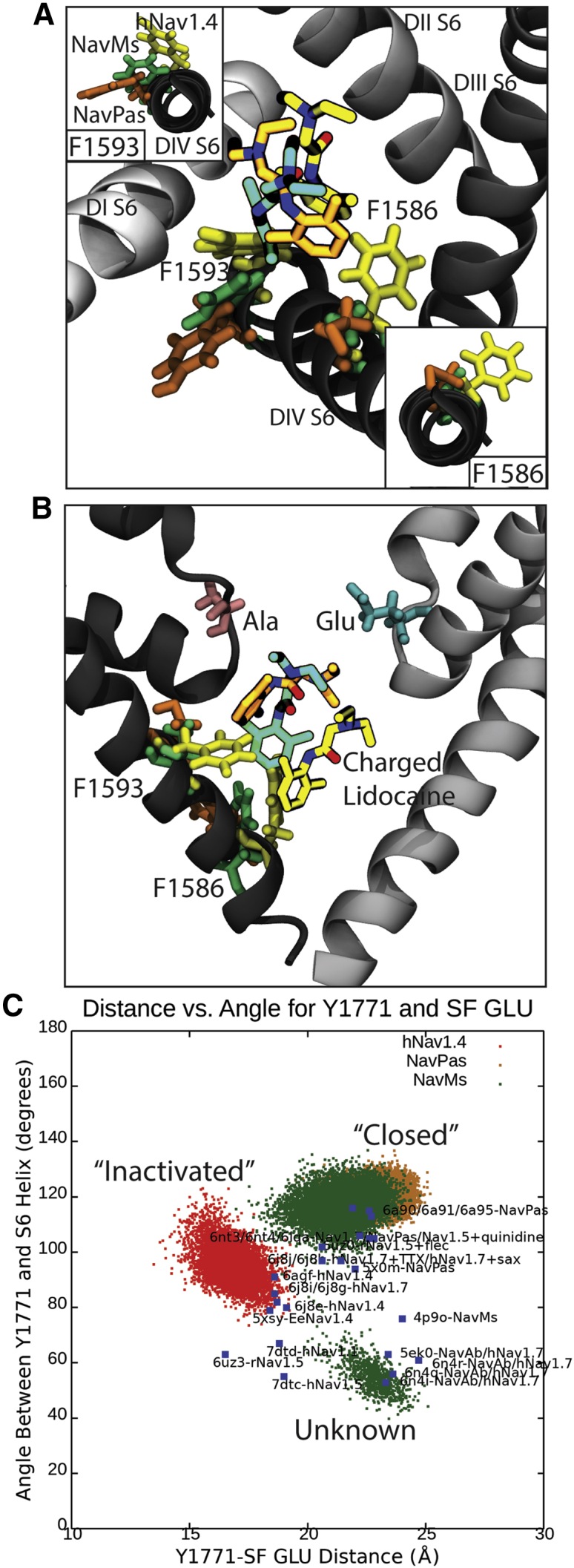
Voltage-gated sodium channels play a vital role in nerve and muscle cells, enabling them to encode and transmit electrical signals. Currently, there exist several classes of drugs that aim to inhibit these channels for therapeutic purposes, including local anesthetics, antiepileptics and antiarrhythmics. However, sodium-channel-inhibiting drugs lack subtype specificity; instead, they inhibit all sodium channels in the human body. Improving understanding of the mechanisms of binding of existing nonselective drugs is important in providing insight into how subtype-selective drugs could be developed. This study used molecular dynamics simulations to investigate the binding of the antiepileptics carbamazepine and lamotrigine and the local anesthetic lidocaine in neutral and charged states to the recently resolved human Nav1.4 channel. Replica exchange solute tempering was used to enable greater sampling of each compound within the pore. It was found that all four compounds show similarities in their binding sites within the pore. However, the positions of the carbamazepine and lamotrigine did not occlude the center of the pore but preferentially bound to homologous domain DII and DIII. The charged and neutral forms of lidocaine positioned themselves more centrally in the pore, with more common interactions with DIV. The best localized binding site was for charged lidocaine, whose aromatic moiety interacted with Y1593, whereas the amine projected toward the selectivity filter. Comparisons with our previous simulations and published structures highlight potential differences between tonic and use-dependent block related to conformational changes occurring in the pore.
Copyright © 2021 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Andavan G.S., Lemmens-Gruber R. Voltage-gated sodium channels: mutations, channelopathies and targets. Curr. Med. Chem. 2011;18:377–397. - PubMed
-
- Shen H., Zhou Q., et al. Yan N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science. 2017;355:eaal4326. - PubMed
-
- Yan Z., Zhou Q., et al. Yan N. Structure of the Nav1.4-β1 complex from electric eel. Cell. 2017;170:470–482.e11. - PubMed
-
- Pan X., Li Z., et al. Yan N. Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science. 2018;362:eaau2486. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
