

Modelling the active SARS-CoV-2 helicase complex as a basis for structure-based inhibitor design
- PMID: 34777769
- PMCID: PMC8528070
- DOI: 10.1039/d1sc02775a
Modelling the active SARS-CoV-2 helicase complex as a basis for structure-based inhibitor design
Abstract
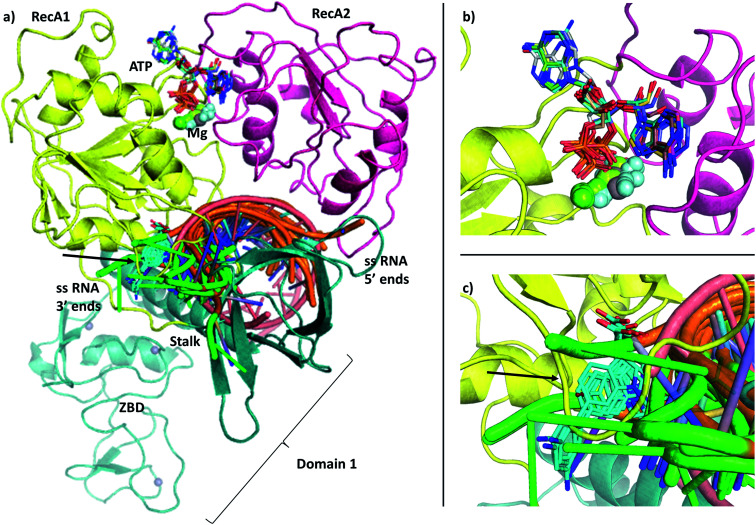
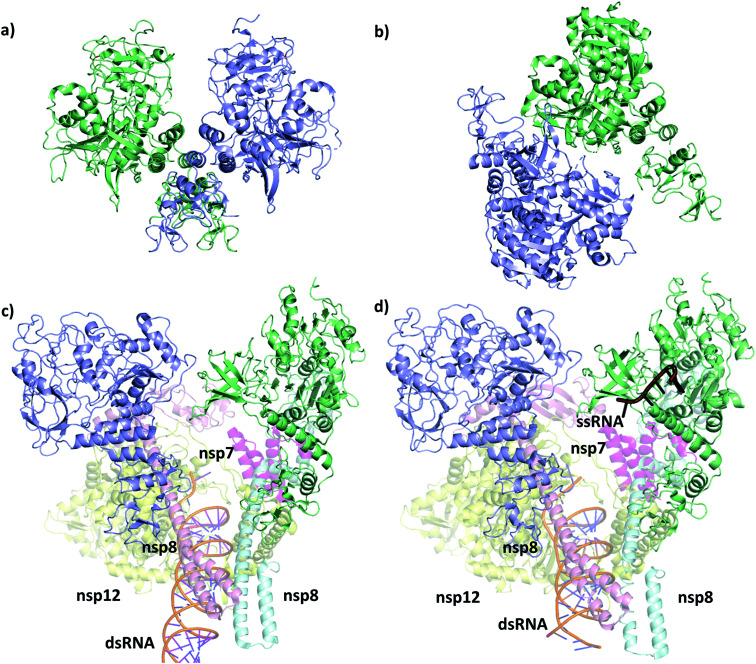
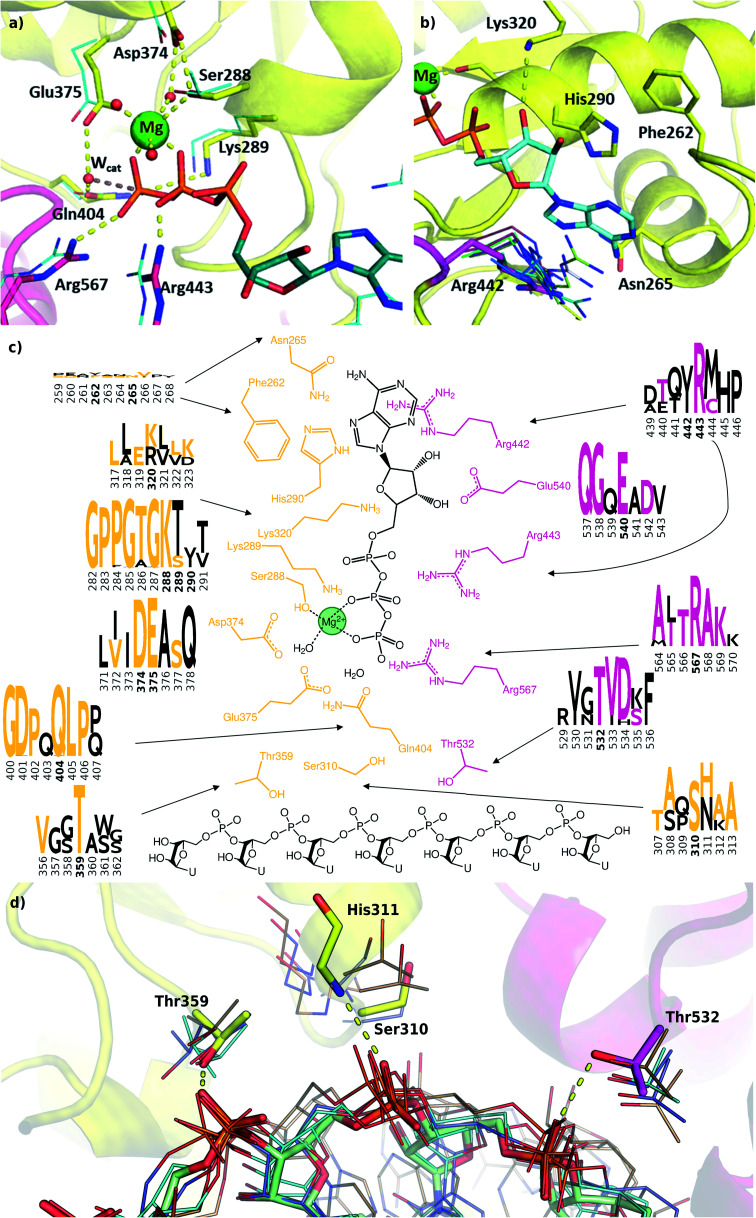
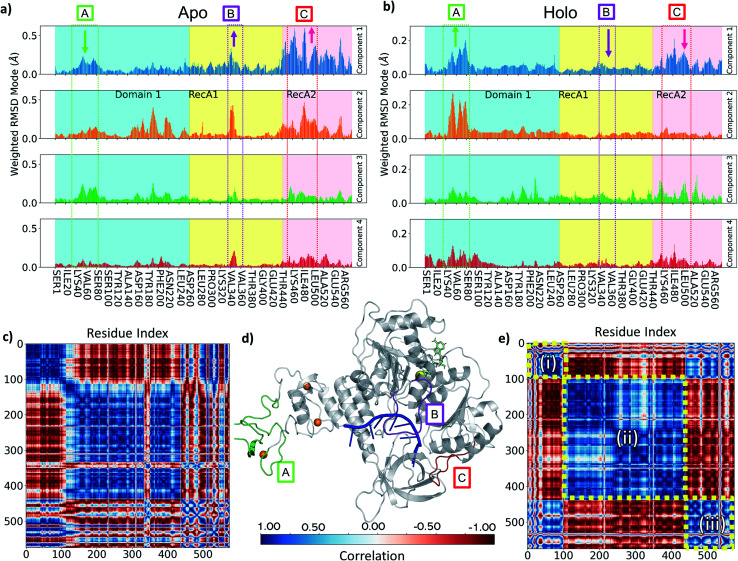
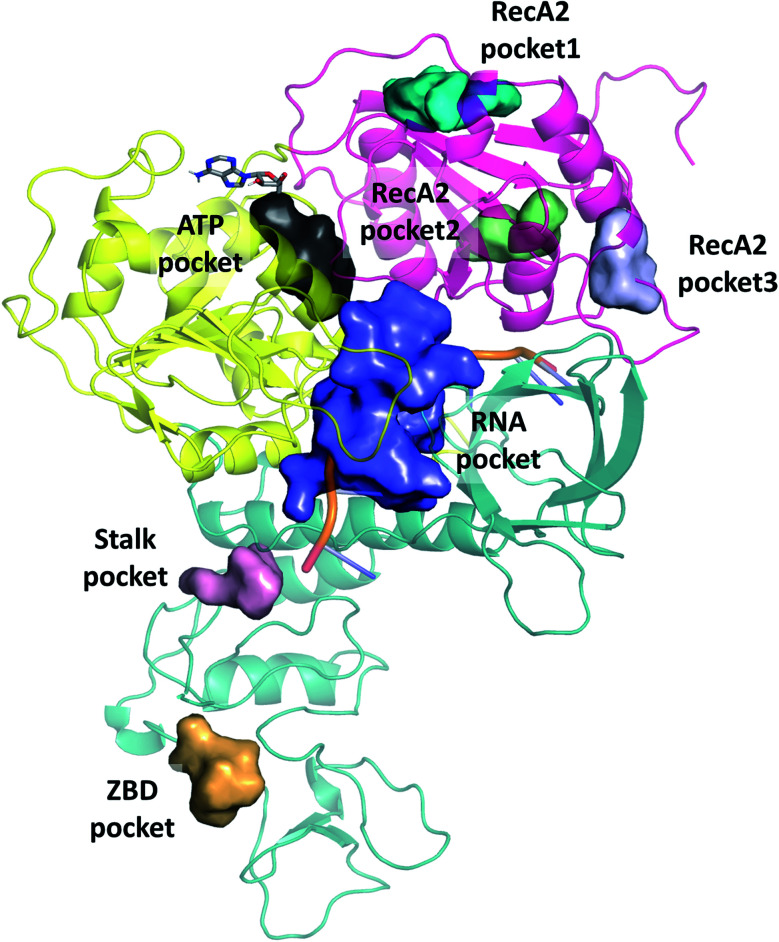
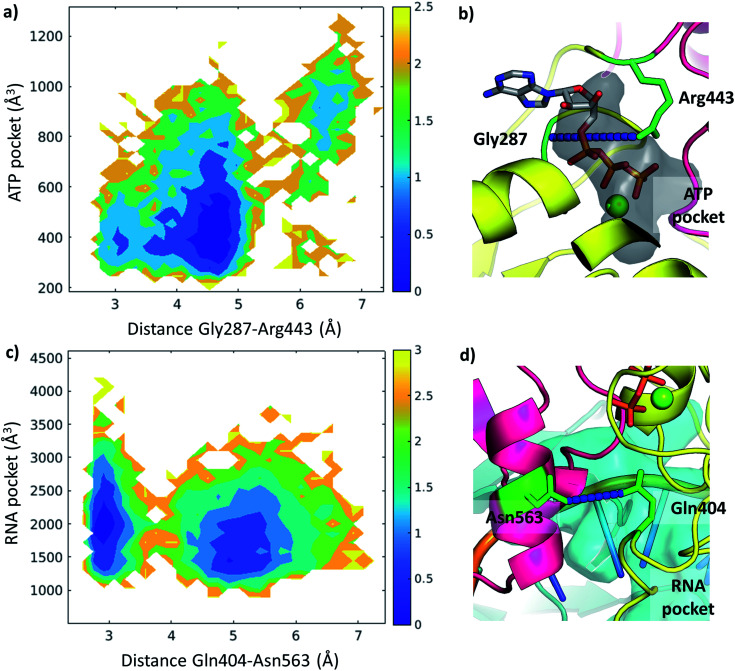
The RNA helicase (non-structural protein 13, NSP13) of SARS-CoV-2 is essential for viral replication, and it is highly conserved among the coronaviridae family, thus a prominent drug target to treat COVID-19. We present here structural models and dynamics of the helicase in complex with its native substrates based on thorough analysis of homologous sequences and existing experimental structures. We performed and analysed microseconds of molecular dynamics (MD) simulations, and our model provides valuable insights to the binding of the ATP and ssRNA at the atomic level. We identify the principal motions characterising the enzyme and highlight the effect of the natural substrates on this dynamics. Furthermore, allosteric binding sites are suggested by our pocket analysis. Our obtained structural and dynamical insights are important for subsequent studies of the catalytic function and for the development of specific inhibitors at our characterised binding pockets for this promising COVID-19 drug target.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures


References
-
- Beigel J. H. Tomashek K. M. Dodd L. E. Mehta A. K. Zingman B. S. Kalil A. C. Hohmann E. Chu H. Y. Luetkemeyer A. Kline S. Lopez de Castilla D. Finberg R. W. Dierberg K. Tapson V. Hsieh L. Patterson T. F. Paredes R. Sweeney D. A. Short W. R. Touloumi G. Lye D. C. Ohmagari N. Oh M. Ruiz-Palacios G. M. Benfield T. Fätkenheuer G. Kortepeter M. G. Atmar R. L. Creech C. B. Lundgren J. Babiker A. G. Pett S. Neaton J. D. Burgess T. H. Bonnett T. Green M. Makowski M. Osinusi A. Nayak S. Lane H. C. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. - DOI - PMC - PubMed
-
- Wang Y. Zhang D. Du G. Du R. Zhao J. Jin Y. Fu S. Gao L. Cheng Z. Lu Q. Hu Y. Luo G. Wang K. Lu Y. Li H. Wang S. Ruan S. Yang C. Mei C. Wang Y. Ding D. Wu F. Tang X. Ye X. Ye Y. Liu B. Yang J. Yin W. Wang A. Fan G. Zhou F. Liu Z. Gu X. Xu J. Shang L. Zhang Y. Cao L. Guo T. Wan Y. Qin H. Jiang Y. Jaki T. Hayden F. G. Horby P. W. Cao B. Wang C. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous