

Syndecans and Their Synstatins: Targeting an Organizer of Receptor Tyrosine Kinase Signaling at the Cell-Matrix Interface
- PMID: 34778093
- PMCID: PMC8578902
- DOI: 10.3389/fonc.2021.775349
Syndecans and Their Synstatins: Targeting an Organizer of Receptor Tyrosine Kinase Signaling at the Cell-Matrix Interface
Abstract
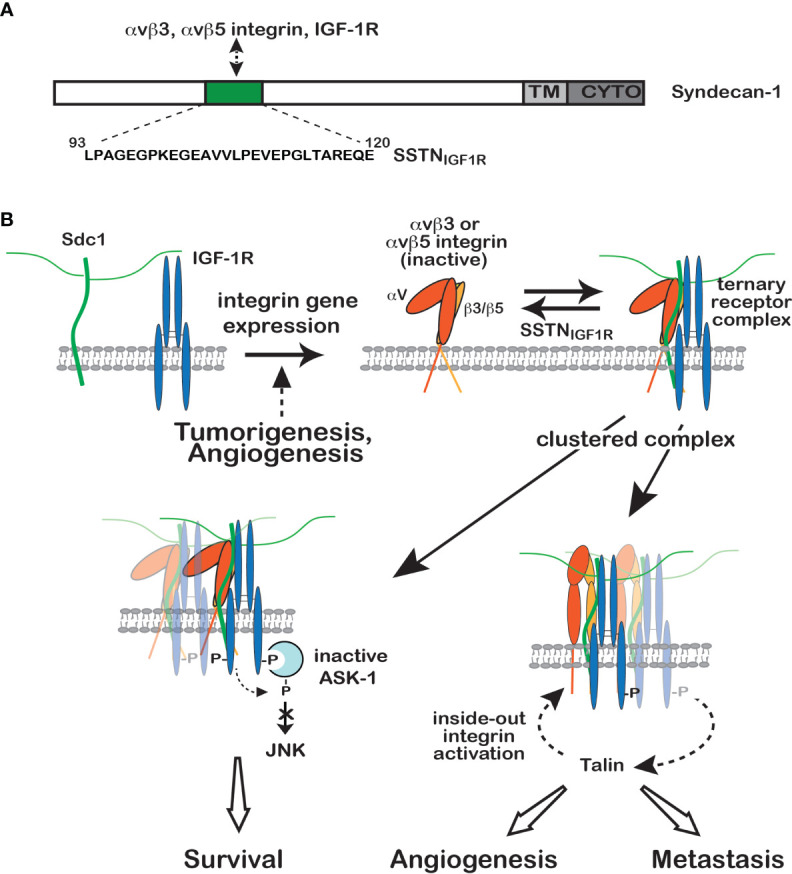
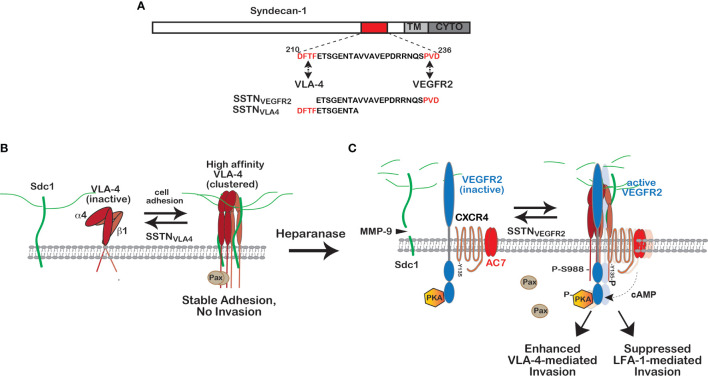
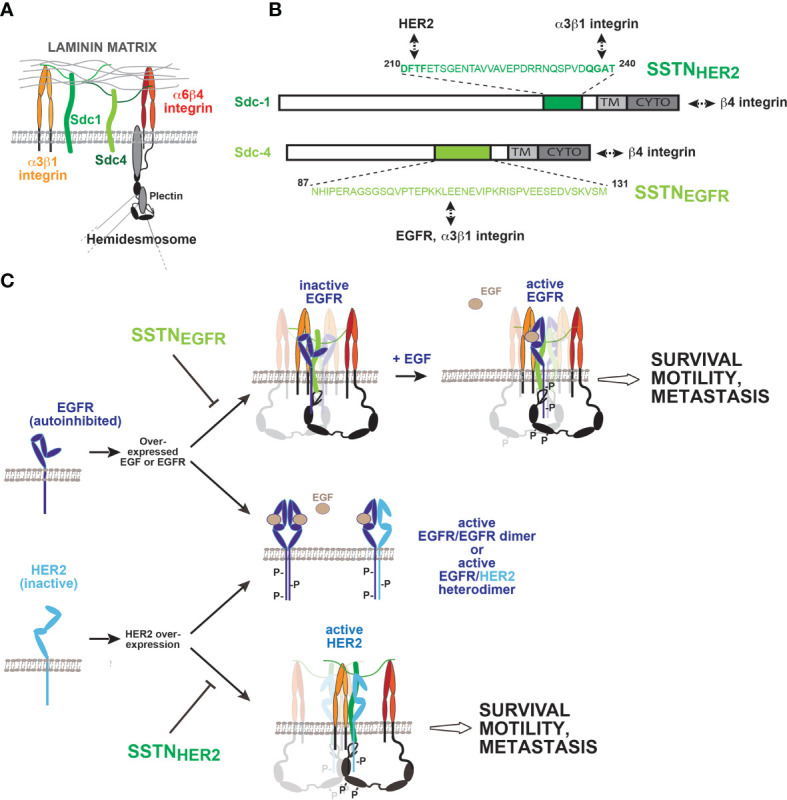
Receptor tyrosine kinases (RTKs) and integrin matrix receptors have well-established roles in tumor cell proliferation, invasion and survival, often functioning in a coordinated fashion at sites of cell-matrix adhesion. Central to this coordination are syndecans, another class of matrix receptor, that organize RTKs and integrins into functional units, relying on docking motifs in the syndecan extracellular domains to capture and localize RTKs (e.g., EGFR, IGF-1R, VEGFR2, HER2) and integrins (e.g., αvβ3, αvβ5, α4β1, α3β1, α6β4) to sites of adhesion. Peptide mimetics of the docking motifs in the syndecans, called "synstatins", prevent assembly of these receptor complexes, block their signaling activities and are highly effective against tumor cell invasion and survival and angiogenesis. This review describes our current understanding of these four syndecan-coupled mechanisms and their inhibitory synstatins (SSTNIGF1R, SSTNVEGFR2, SSTNVLA-4, SSTNEGFR and SSTNHER2).
Keywords: ASK-1; CXCR4; EGFR; IGF-1R; VEGFR2; integrin signaling; synstatin.
Copyright © 2021 Rapraeger.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Syndecan-1 and Syndecan-4 Capture Epidermal Growth Factor Receptor Family Members and the α3β1 Integrin Via Binding Sites in Their Ectodomains: NOVEL SYNSTATINS PREVENT KINASE CAPTURE AND INHIBIT α6β4-INTEGRIN-DEPENDENT EPITHELIAL CELL MOTILITY.J Biol Chem. 2015 Oct 23;290(43):26103-13. doi: 10.1074/jbc.M115.679084. Epub 2015 Sep 8. J Biol Chem. 2015. PMID: 26350464 Free PMC article.
-
Synstatin: a selective inhibitor of the syndecan-1-coupled IGF1R-αvβ3 integrin complex in tumorigenesis and angiogenesis.FEBS J. 2013 May;280(10):2207-15. doi: 10.1111/febs.12160. Epub 2013 Feb 24. FEBS J. 2013. PMID: 23375101 Free PMC article. Review.
-
Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with α6β4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival.J Biol Chem. 2014 Oct 31;289(44):30318-30332. doi: 10.1074/jbc.M114.586438. Epub 2014 Sep 8. J Biol Chem. 2014. PMID: 25202019 Free PMC article.
-
Plasma membrane proteoglycans syndecan-2 and syndecan-4 engage with EGFR and RON kinase to sustain carcinoma cell cycle progression.J Biol Chem. 2022 Jun;298(6):102029. doi: 10.1016/j.jbc.2022.102029. Epub 2022 May 13. J Biol Chem. 2022. PMID: 35569509 Free PMC article.
-
Syndecans in tumor cell adhesion and signaling.Reprod Biol Endocrinol. 2004 Jan 7;2:3. doi: 10.1186/1477-7827-2-3. Reprod Biol Endocrinol. 2004. PMID: 14711376 Free PMC article. Review.
Cited by
-
Targeted therapy for multiple myeloma: an overview on CD138-based strategies.Front Oncol. 2024 Apr 9;14:1370854. doi: 10.3389/fonc.2024.1370854. eCollection 2024. Front Oncol. 2024. PMID: 38655136 Free PMC article. Review.
-
Signaling Pathways Associated with Chronic Wound Progression: A Systems Biology Approach.Antioxidants (Basel). 2022 Jul 31;11(8):1506. doi: 10.3390/antiox11081506. Antioxidants (Basel). 2022. PMID: 36009225 Free PMC article.
-
Exploring New Frontiers: Alternative Breast Cancer Treatments Through Glycocalyx Research.Breast J. 2025 May 22;2025:9952727. doi: 10.1155/tbj/9952727. eCollection 2025. Breast J. 2025. PMID: 40443562 Free PMC article. Review.
-
Fibrotic lung ECM upregulates SDC4/integrin-αvβ1 interaction and the interfering peptide SDC487-131 and its derivative peptides alleviate pulmonary fibrosis.Regen Biomater. 2025 Jun 16;12:rbaf057. doi: 10.1093/rb/rbaf057. eCollection 2025. Regen Biomater. 2025. PMID: 40747330 Free PMC article.
-
Thy-1 (CD90)-regulated cell adhesion and migration of mesenchymal cells: insights into adhesomes, mechanical forces, and signaling pathways.Front Cell Dev Biol. 2023 Nov 30;11:1221306. doi: 10.3389/fcell.2023.1221306. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 38099295 Free PMC article. Review.
References
-
- Miyamoto S, Teramoto H, Gutkind JS, Yamada KM. Integrins can Collaborate With Growth Factors for Phosphorylation of Receptor Tyrosine Kinases and MAP Kinase Activation: Roles of Integrin Aggregation and Occupancy of Receptors. J Cell Biol (1996) 135:1633–42. doi: 10.1083/jcb.135.6.1633 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous