

Aberrant Extrafollicular B Cells, Immune Dysfunction, Myeloid Inflammation, and MyD88-Mutant Progenitors Precede Waldenstrom Macroglobulinemia
- PMID: 34778800
- PMCID: PMC8580616
- DOI: 10.1158/2643-3230.BCD-21-0043
Aberrant Extrafollicular B Cells, Immune Dysfunction, Myeloid Inflammation, and MyD88-Mutant Progenitors Precede Waldenstrom Macroglobulinemia
Abstract
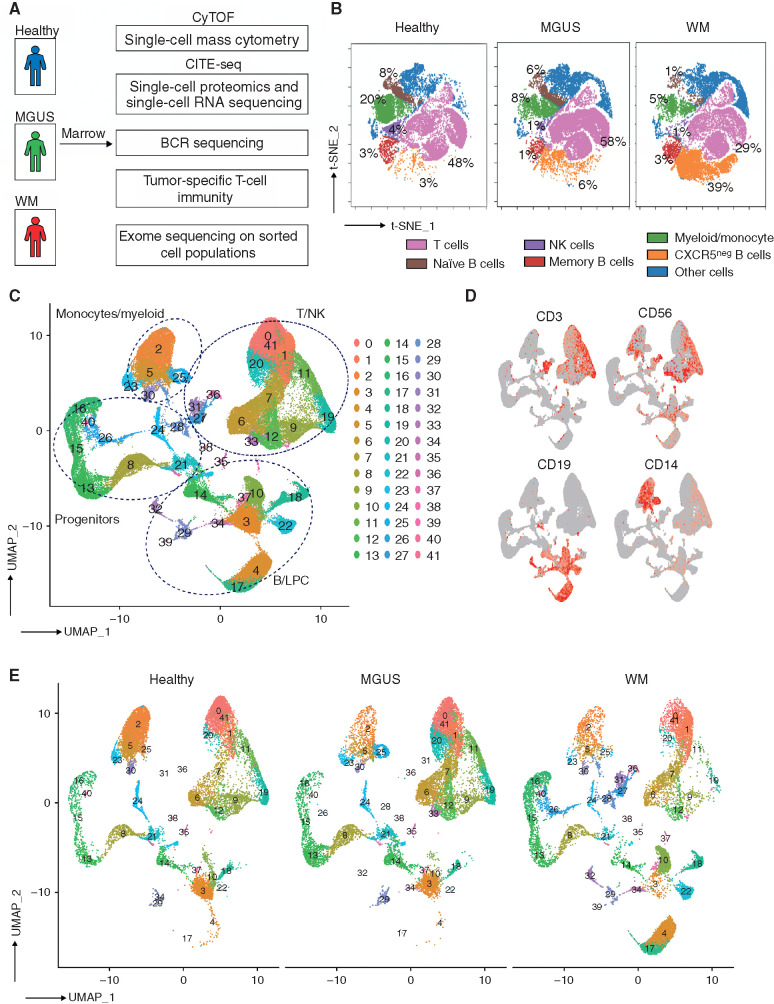
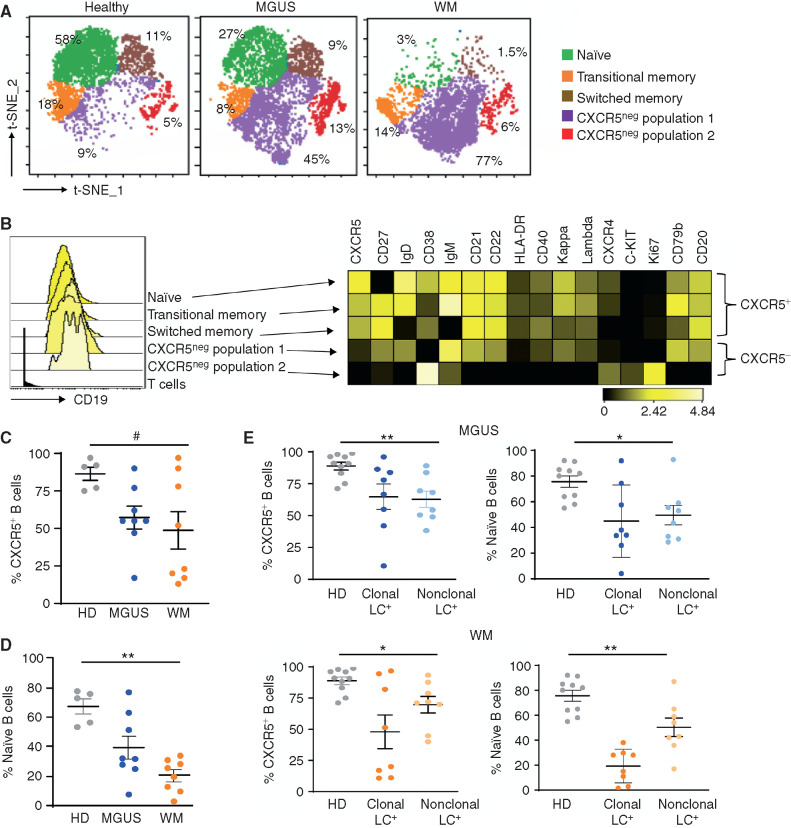
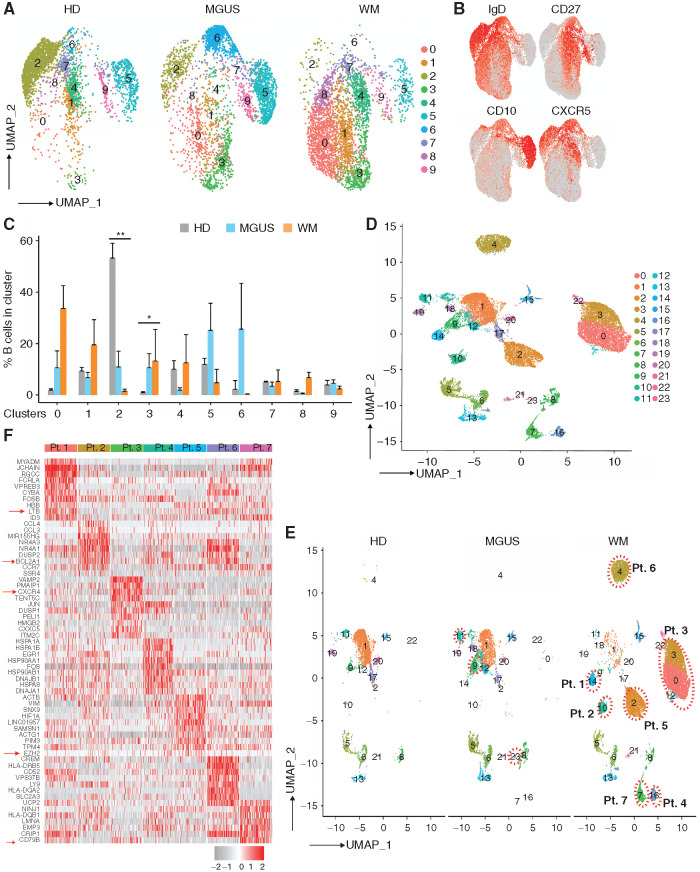
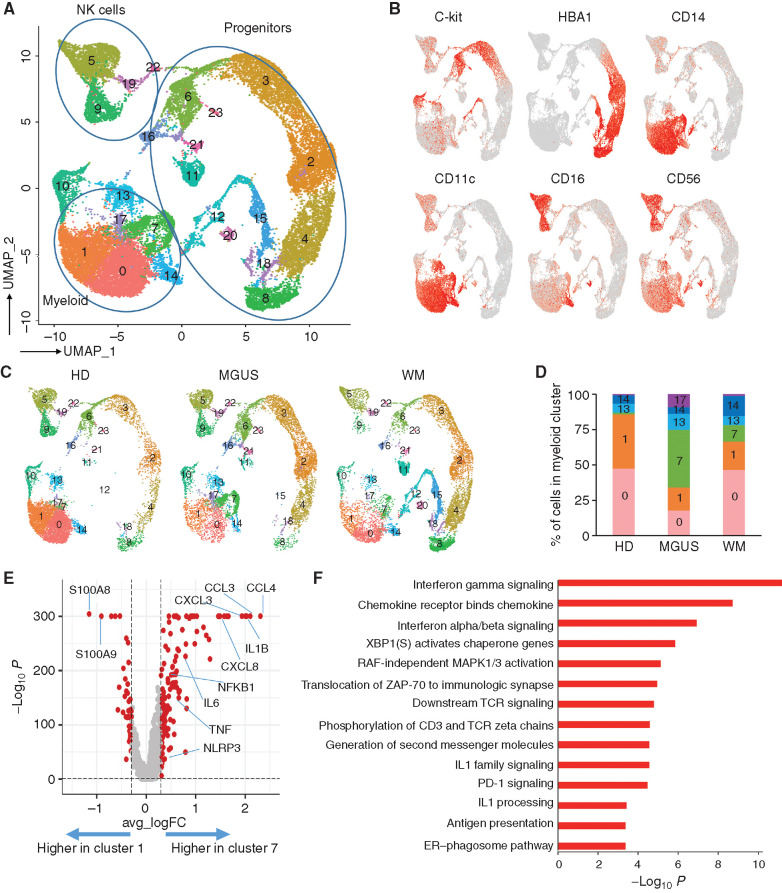
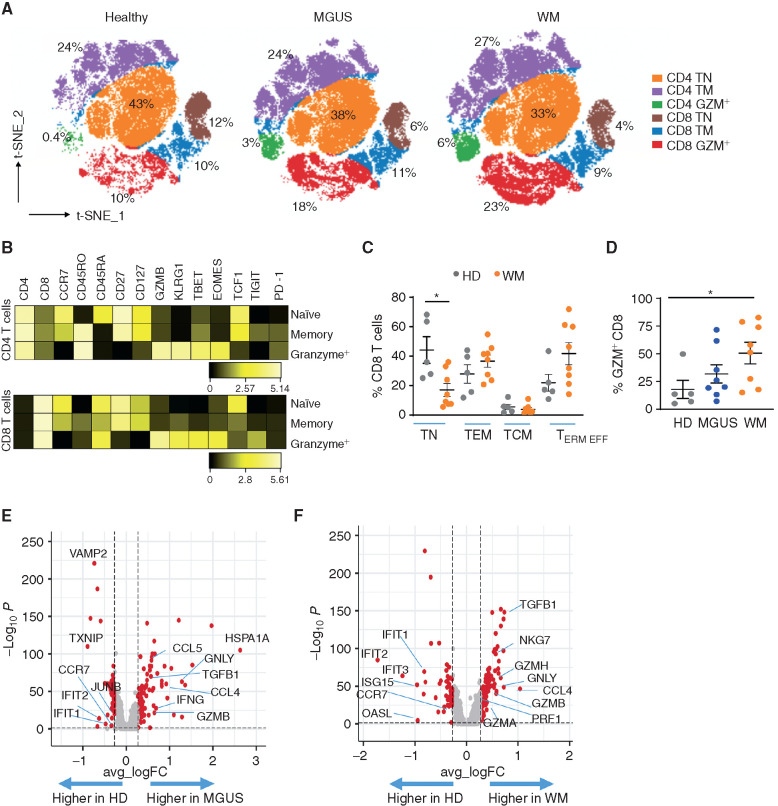
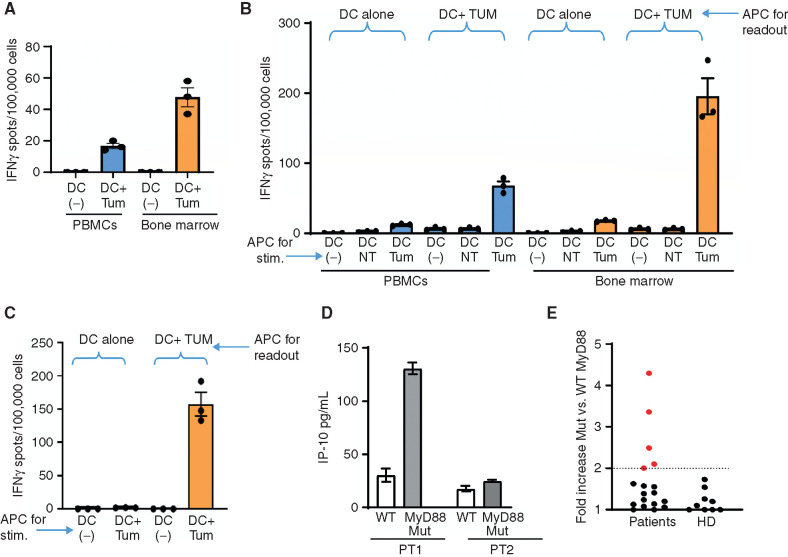
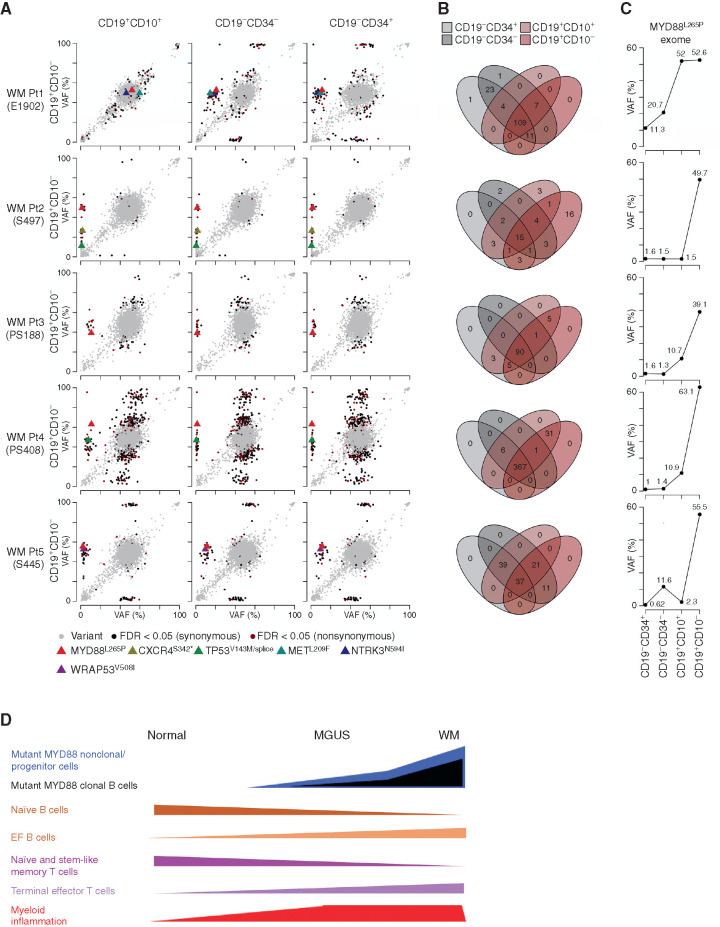
Waldenstrom macroglobulinemia (WM) and its precursor IgM gammopathy are distinct disorders characterized by clonal mature IgM-expressing B-cell outgrowth in the bone marrow. Here, we show by high-dimensional single-cell immunogenomic profiling of patient samples that these disorders originate in the setting of global B-cell compartment alterations, characterized by expansion of genomically aberrant extrafollicular B cells of the nonmalignant clonotype. Alterations in the immune microenvironment preceding malignant clonal expansion include myeloid inflammation and naïve B- and T-cell depletion. Host response to these early lesions involves clone-specific T-cell immunity that may include MYD88 mutation-specific responses. Hematopoietic progenitors carry the oncogenic MYD88 mutations characteristic of the malignant WM clone. These data support a model for WM pathogenesis wherein oncogenic alterations and signaling in progenitors, myeloid inflammation, and global alterations in extrafollicular B cells create the milieu promoting extranodal pattern of growth in differentiated malignant cells.
Significance: These data provide evidence that growth of the malignant clone in WM is preceded by expansion of extrafollicular B cells, myeloid inflammation, and immune dysfunction in the preneoplastic phase. These changes may be related in part to MYD88 oncogenic signaling in pre-B progenitor cells and suggest a novel model for WM pathogenesis. This article is highlighted in the In This Issue feature, p. 549.
©2021 American Association for Cancer Research.
Figures
Similar articles
-
Genomic and immune profiling of prognostic risk groups in IgM gammopathy reveals novel biomarkers beyond MYD88 L265P.Front Immunol. 2025 Jul 1;16:1604089. doi: 10.3389/fimmu.2025.1604089. eCollection 2025. Front Immunol. 2025. PMID: 40666506 Free PMC article.
-
Expanded tumor-associated polymorphonuclear myeloid-derived suppressor cells in Waldenstrom macroglobulinemia display immune suppressive activity.Blood Cancer J. 2024 Dec 18;14(1):217. doi: 10.1038/s41408-024-01173-w. Blood Cancer J. 2024. PMID: 39695096 Free PMC article.
-
GLI3 Is Required for M2 Macrophage Polarization and M2-Mediated Waldenström Macroglobulinemia Growth and Survival.Int J Mol Sci. 2024 Dec 6;25(23):13120. doi: 10.3390/ijms252313120. Int J Mol Sci. 2024. PMID: 39684827 Free PMC article.
-
Report of Consensus Panel 6 from the 12th International Workshop on Waldenstrom's Macroglobulinemia on Diagnosis and Management of Transformed Waldenstrom's Macroglobulinemia.Semin Hematol. 2025 Apr;62(2):120-125. doi: 10.1053/j.seminhematol.2025.04.003. Epub 2025 Apr 8. Semin Hematol. 2025. PMID: 40382198
-
Report of Consensus Panel 4 from the 12th International Workshop on Waldenstrom's Macroglobulinemia on the management of patients with non-IgM lymphoplasmacytic lymphoma.Semin Hematol. 2025 Apr;62(2):106-112. doi: 10.1053/j.seminhematol.2025.04.002. Epub 2025 Apr 8. Semin Hematol. 2025. PMID: 40382197
Cited by
-
Genomic and immune profiling of prognostic risk groups in IgM gammopathy reveals novel biomarkers beyond MYD88 L265P.Front Immunol. 2025 Jul 1;16:1604089. doi: 10.3389/fimmu.2025.1604089. eCollection 2025. Front Immunol. 2025. PMID: 40666506 Free PMC article.
-
Prognostic impact of MYD88 and CXCR4 mutations assessed by droplet digital polymerase chain reaction in IgM monoclonal gammopathy of undetermined significance and smouldering Waldenström macroglobulinaemia.Br J Haematol. 2023 Jan;200(2):187-196. doi: 10.1111/bjh.18502. Epub 2022 Oct 9. Br J Haematol. 2023. PMID: 36210485 Free PMC article.
-
Association of race/ethnicity with innate immune tumor microenvironment of children with B-acute lymphoblastic leukemia.J Immunother Cancer. 2022 Jun;10(6):e004774. doi: 10.1136/jitc-2022-004774. J Immunother Cancer. 2022. PMID: 35710294 Free PMC article.
-
Changes in Bone Marrow Tumor and Immune Cells Correlate with Durability of Remissions Following BCMA CAR T Therapy in Myeloma.Blood Cancer Discov. 2022 Nov 2;3(6):490-501. doi: 10.1158/2643-3230.BCD-22-0018. Blood Cancer Discov. 2022. PMID: 36026513 Free PMC article.
-
The immune system in multiple myeloma and precursor states: Lessons and implications for immunotherapy and interception.Am J Hematol. 2023 Mar;98 Suppl 2(Suppl 2):S4-S12. doi: 10.1002/ajh.26752. Epub 2022 Oct 24. Am J Hematol. 2023. PMID: 36194782 Free PMC article. Review.
References
-
- Gertz MA.Waldenstrom macroglobulinemia: 2020 update on diagnosis, risk stratification, and management. Am J Hematol 2021;96:258–69. - PubMed
-
- Dhodapkar MV, Jacobson JL, Gertz MA, Rivkin SE, Roodman GD, Tuscano JM, et al. Prognostic factors and response to fludarabine therapy in patients with Waldenstrom macroglobulinemia: results of United States intergroup trial (Southwest Oncology Group S9003). Blood 2001;98:41–8. - PubMed
-
- Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med 2012;367:826–33. - PubMed
-
- Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013;121:2051–8. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
