

Distinct epilepsy phenotypes and response to drugs in KCNA1 gain- and loss-of function variants
- PMID: 34778950
- PMCID: PMC9299230
- DOI: 10.1111/epi.17118
Distinct epilepsy phenotypes and response to drugs in KCNA1 gain- and loss-of function variants
Abstract
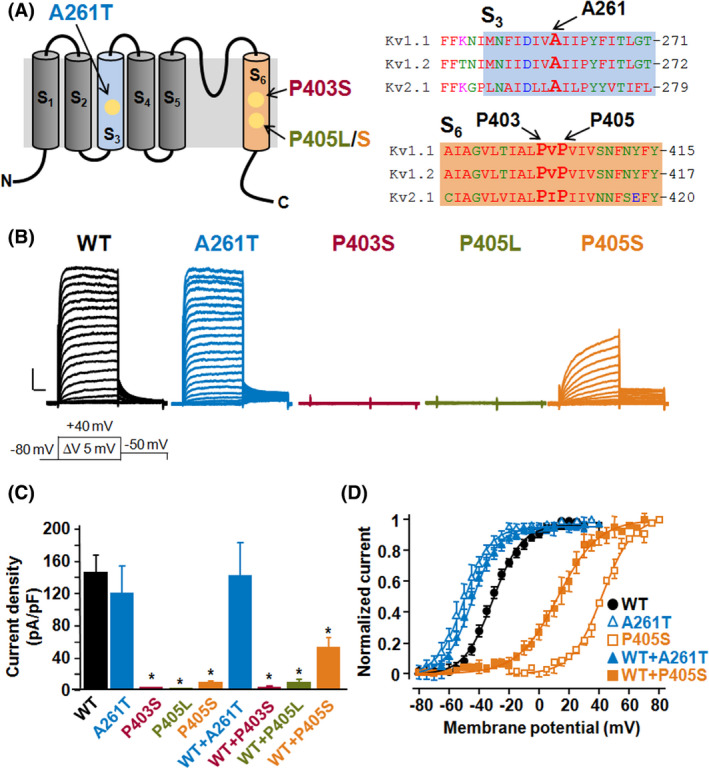
A wide phenotypic spectrum of neurological diseases is associated with KCNA1 (Kv1.1) variants. To investigate the molecular basis of such a heterogeneous clinical presentation and identify the possible correlation with in vitro phenotypes, we compared the functional consequences of three heterozygous de novo variants (p.P403S, p.P405L, and p.P405S) in Kv1.1 pore region found in four patients with severe developmental and epileptic encephalopathy (DEE), with those of a de novo variant in the voltage sensor (p.A261T) identified in two patients with mild, carbamazepine-responsive, focal epilepsy. Patch-clamp electrophysiology was used to investigate the functional properties of mutant Kv1.1 subunits, both expressed as homomers and heteromers with wild-type Kv1.1 subunits. KCNA1 pore mutations markedly decreased (p. P405S) or fully suppressed (p. P403S, p. P405L) Kv1.1-mediated currents, exerting loss-of-function (LoF) effects. By contrast, channels carrying the p.A261T variant exhibited a hyperpolarizing shift of the activation process, consistent with a gain-of-function (GoF) effect. The present results unveil a novel correlation between in vitro phenotype (GoF vs LoF) and clinical course (mild vs severe) in KCNA1-related phenotypes. The excellent clinical response to carbamazepine observed in the patients carrying the A261T variant suggests an exquisite sensitivity of KCNA1 GoF to sodium channel inhibition that should be further explored.
Keywords: KCNA1; developmental encephalopathies; epilepsy; gain-of-function variants; loss-of-function variants; potassium channels.
© 2021 The Authors. Epilepsia published by Wiley Periodicals LLC on behalf of International League Against Epilepsy.
Conflict of interest statement
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Figures
References
-
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–40. - PubMed
-
- Rogers A, Golumbek P, Cellini E, Doccini V, Guerrini R, Wallgren‐Pettersson C, et al. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am J Med Genet A. 2018;176:1748–52. - PubMed
