

Bacterial genomic epidemiology with mixed samples
- PMID: 34779765
- PMCID: PMC8743562
- DOI: 10.1099/mgen.0.000691
Bacterial genomic epidemiology with mixed samples
Abstract
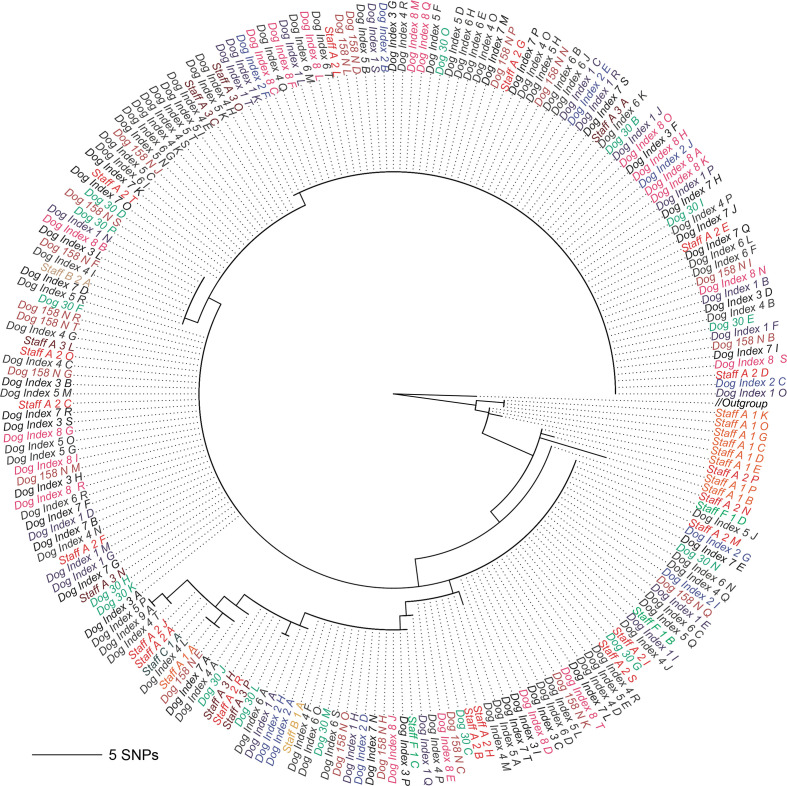
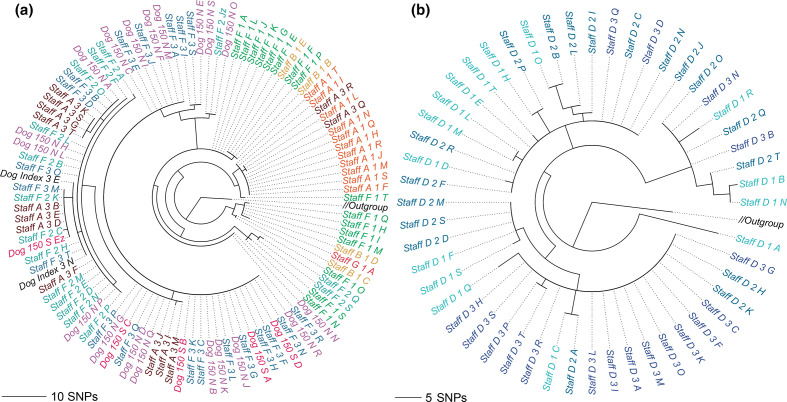
Genomic epidemiology is a tool for tracing transmission of pathogens based on whole-genome sequencing. We introduce the mGEMS pipeline for genomic epidemiology with plate sweeps representing mixed samples of a target pathogen, opening the possibility to sequence all colonies on selective plates with a single DNA extraction and sequencing step. The pipeline includes the novel mGEMS read binner for probabilistic assignments of sequencing reads, and the scalable pseudoaligner Themisto. We demonstrate the effectiveness of our approach using closely related samples in a nosocomial setting, obtaining results that are comparable to those based on single-colony picks. Our results lend firm support to more widespread consideration of genomic epidemiology with mixed infection samples.
Keywords: genomic epidemiology; pathogen surveillance; plate sweeps; probabilistic modelling; pseudoalignment.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources