

Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review
- PMID: 34780255
- PMCID: PMC9034711
- DOI: 10.1161/CIRCULATIONAHA.121.055890
Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review
Abstract
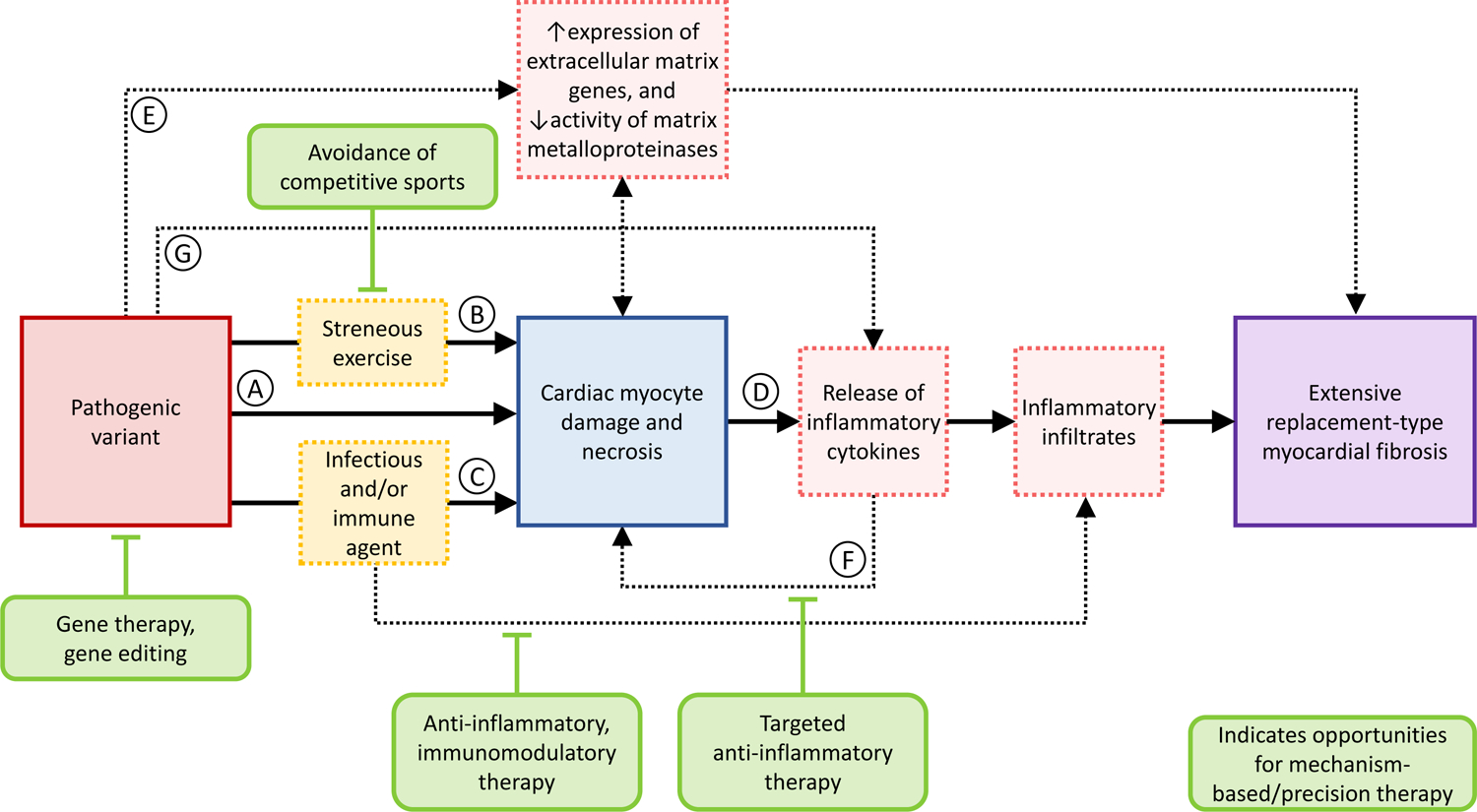
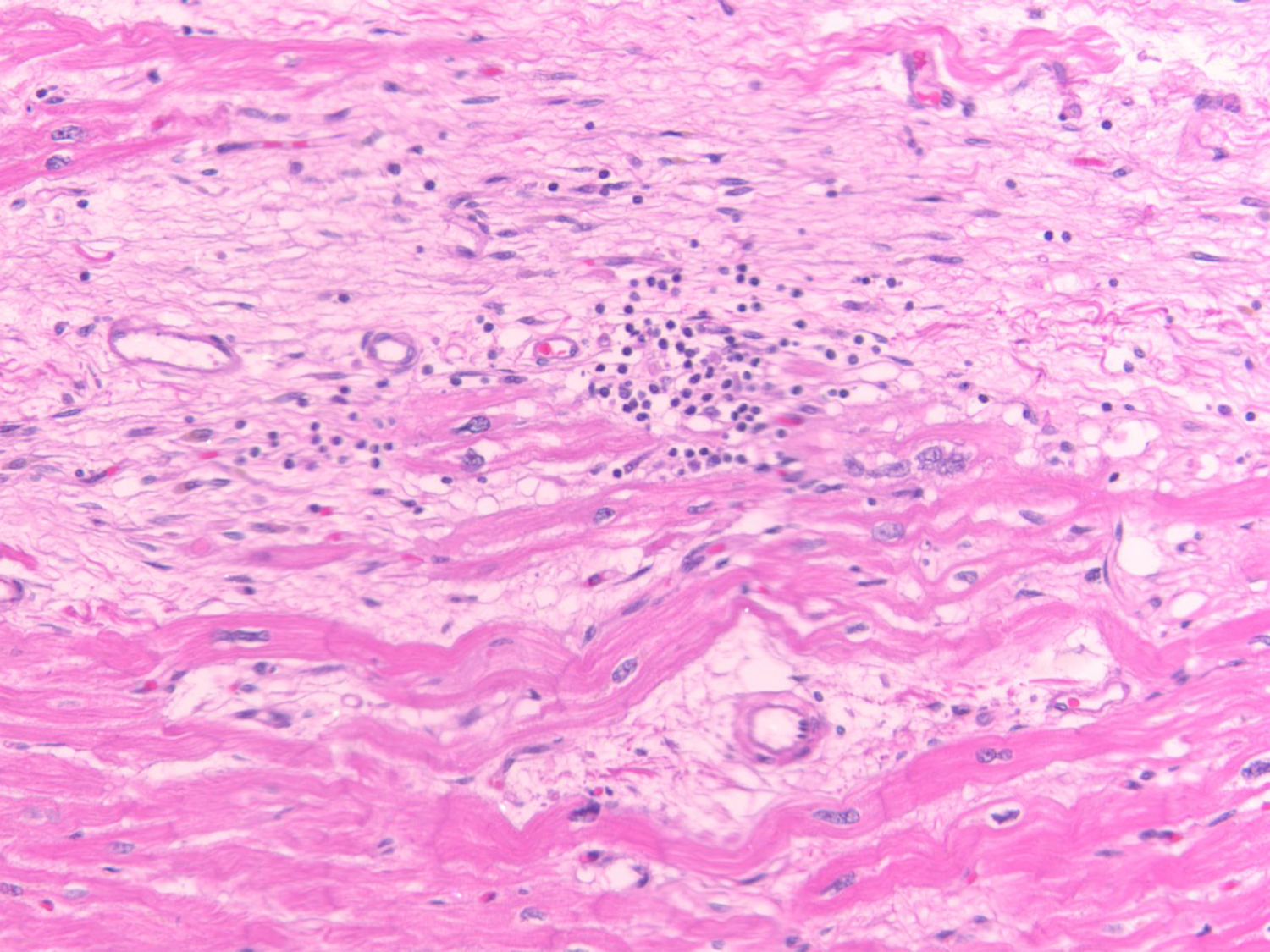
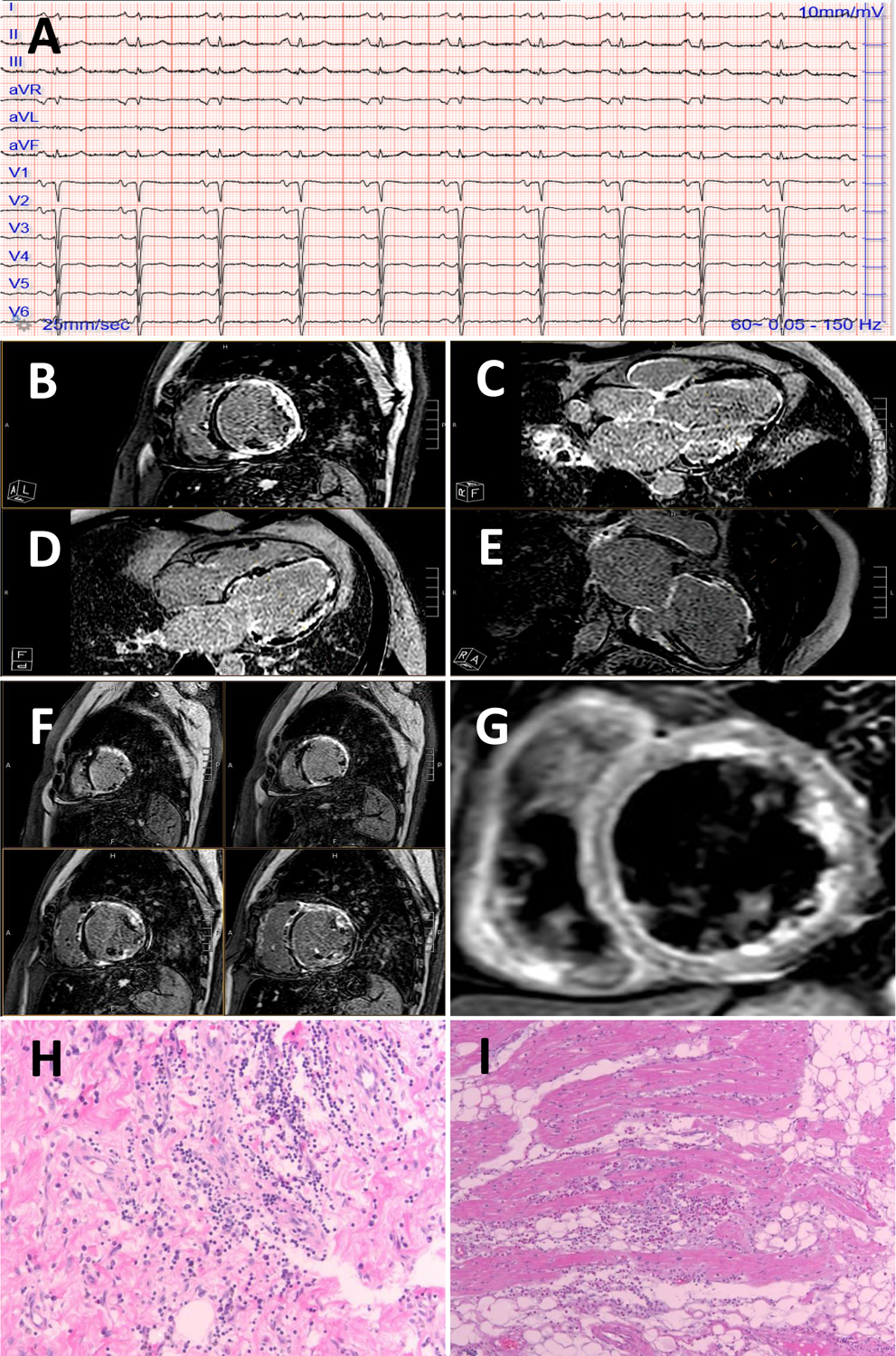
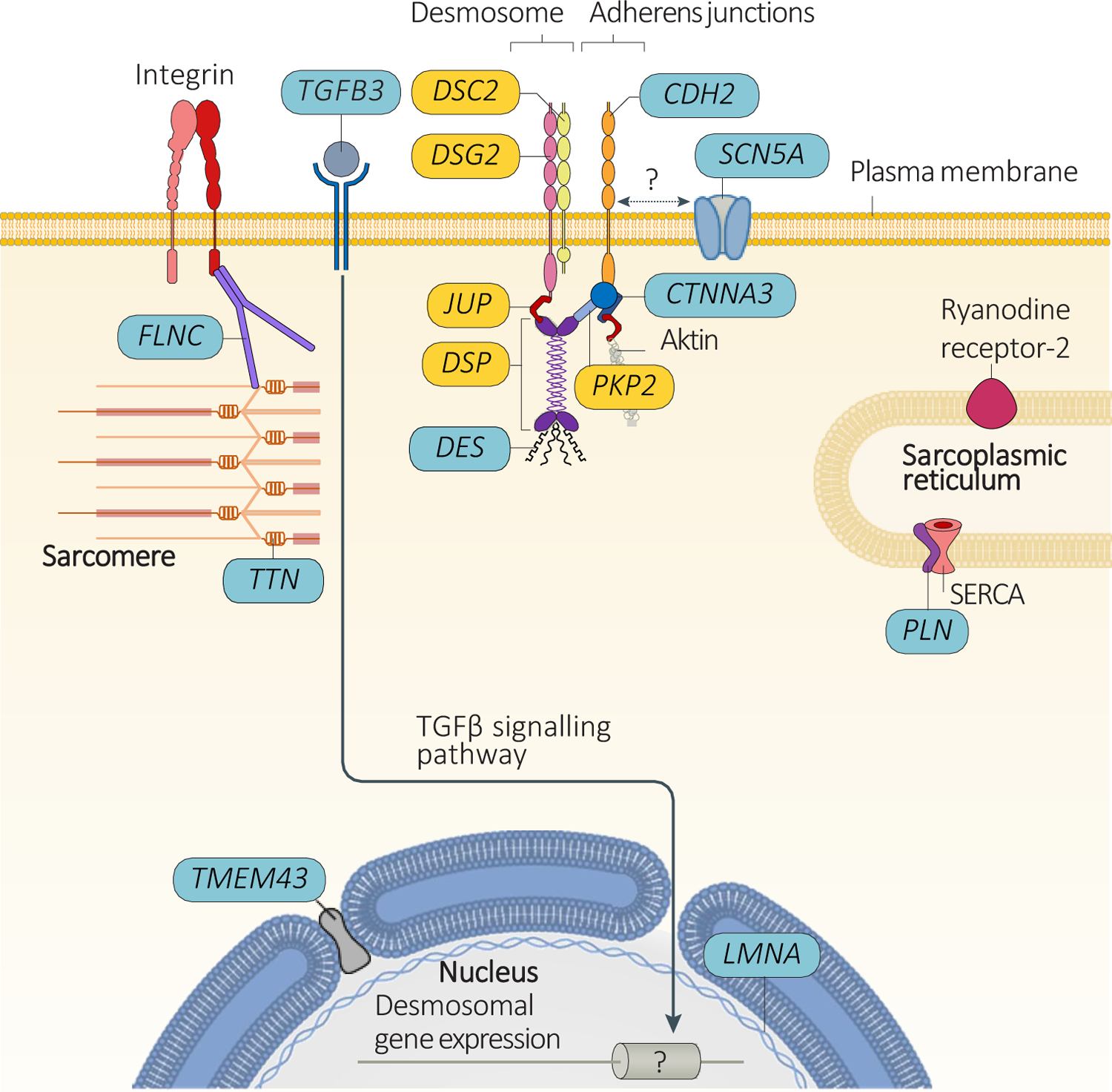
Arrhythmogenic cardiomyopathy (ACM) is a primary disease of the myocardium, predominantly caused by genetic defects in proteins of the cardiac intercalated disc, particularly, desmosomes. Transmission is mostly autosomal dominant with incomplete penetrance. ACM also has wide phenotype variability, ranging from premature ventricular contractions to sudden cardiac death and heart failure. Among other drivers and modulators of phenotype, inflammation in response to viral infection and immune triggers have been postulated to be an aggravator of cardiac myocyte damage and necrosis. This theory is supported by multiple pieces of evidence, including the presence of inflammatory infiltrates in more than two-thirds of ACM hearts, detection of different cardiotropic viruses in sporadic cases of ACM, the fact that patients with ACM often fulfill the histological criteria of active myocarditis, and the abundance of anti-desmoglein-2, antiheart, and anti-intercalated disk autoantibodies in patients with arrhythmogenic right ventricular cardiomyopathy. In keeping with the frequent familial occurrence of ACM, it has been proposed that, in addition to genetic predisposition to progressive myocardial damage, a heritable susceptibility to viral infections and immune reactions may explain familial clustering of ACM. Moreover, considerable in vitro and in vivo evidence implicates activated inflammatory signaling in ACM. Although the role of inflammation/immune response in ACM is not entirely clear, inflammation as a driver of phenotype and a potential target for mechanism-based therapy warrants further research. This review discusses the present evidence supporting the role of inflammatory and immune responses in ACM pathogenesis and proposes opportunities for translational and clinical investigation.
Keywords: arrhythmias, cardiac; arrhythmogenic right ventricular dysplasia; autoimmunity; death, sudden, cardiac; genetics; inflammation; myocarditis.
Figures
References
-
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C and Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. - PubMed
-
- Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli-Ducci C, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41:1414–1429. - PMC - PubMed
-
- Corrado D, Perazzolo Marra M, Zorzi A, Beffagna G, Cipriani A, Lazzari M, Migliore F, Pilichou K, Rampazzo A, Rigato I, et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol. 2020;319:106–114. - PubMed
-
- Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E and McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. - PubMed
-
- Briceno DF, Liang JJ, Shirai Y, Markman TM, Chahal A, Tschabrunn C, Zado E, Hyman MC, Kumareswaran R, Arkles JS, et al. Characterization of Structural Changes in Arrhythmogenic Right Ventricular Cardiomyopathy With Recurrent Ventricular Tachycardia After Ablation: Insights From Repeat Electroanatomic Voltage Mapping. Circ Arrhythm Electrophysiol. 2020;13:e007611. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
