

Direct RNA Nanopore Sequencing of Pseudomonas aeruginosa Clone C Transcriptomes
- PMID: 34780302
- PMCID: PMC8765398
- DOI: 10.1128/JB.00418-21
Direct RNA Nanopore Sequencing of Pseudomonas aeruginosa Clone C Transcriptomes
Abstract
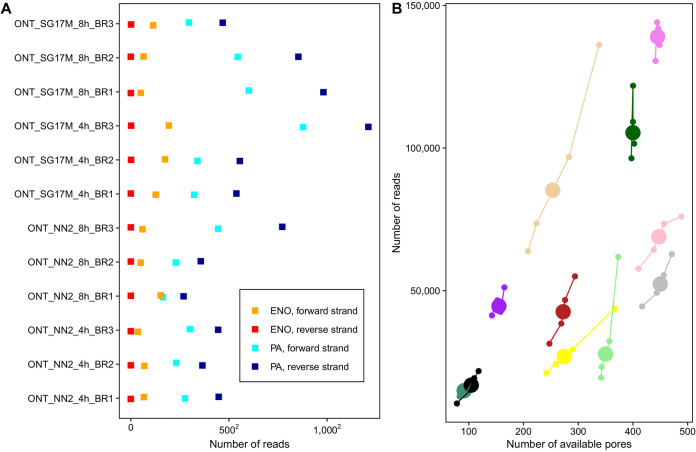
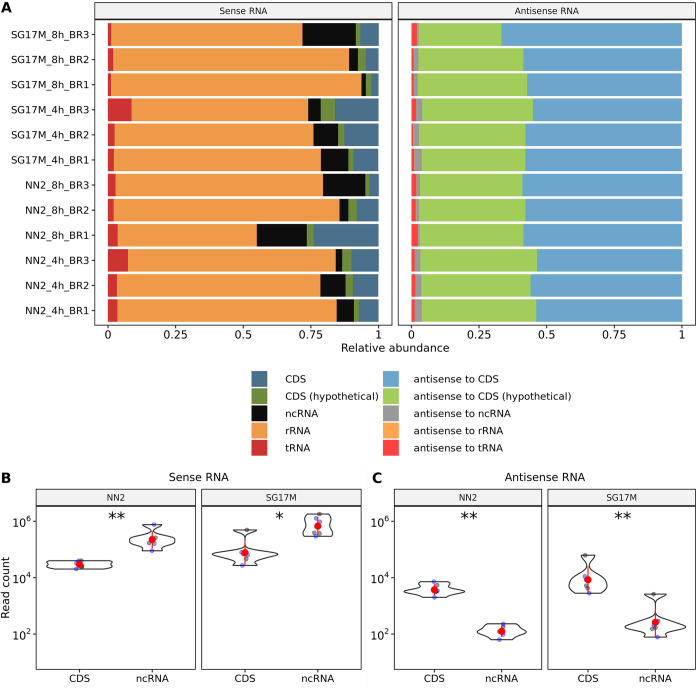
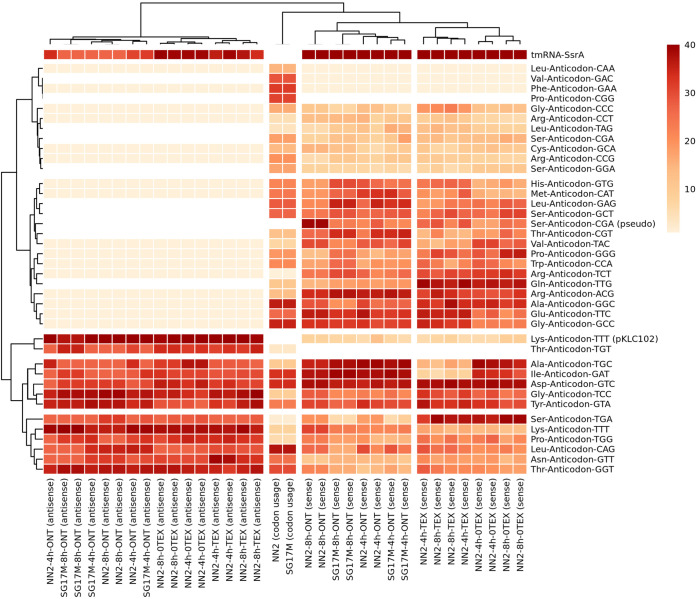
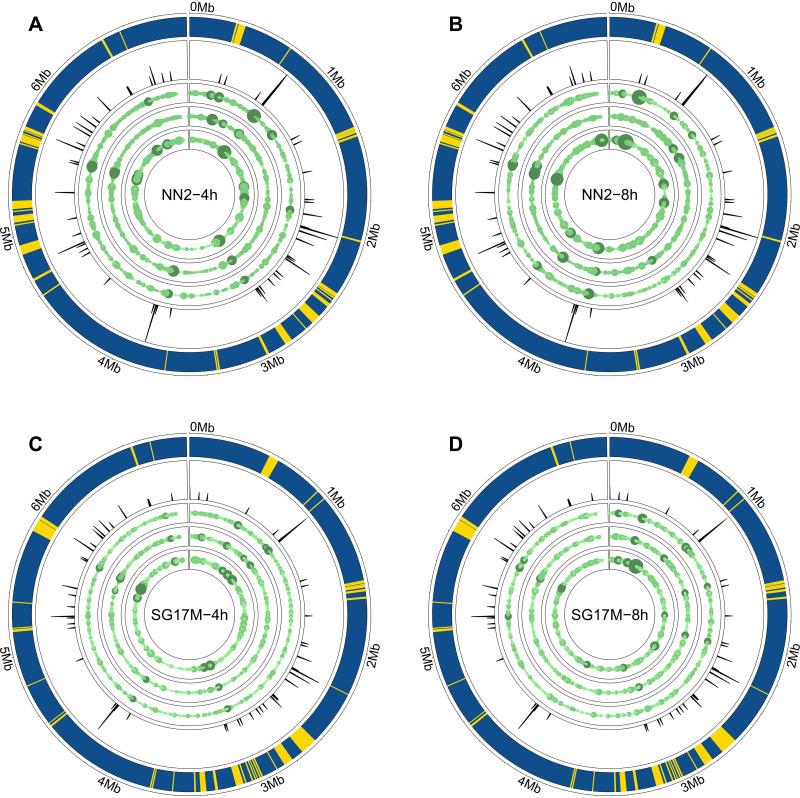
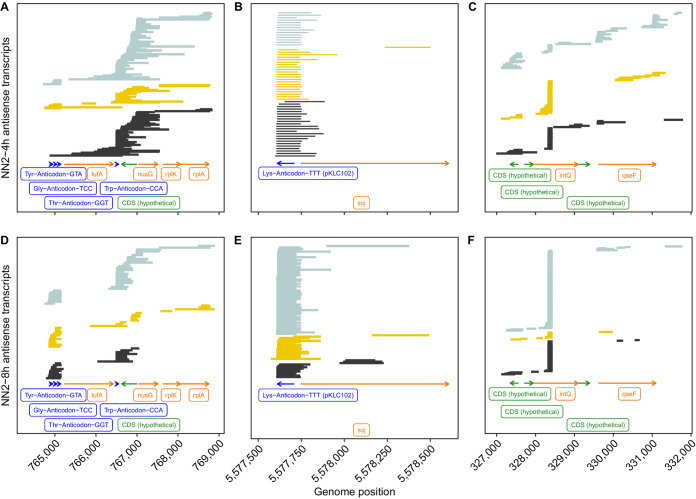
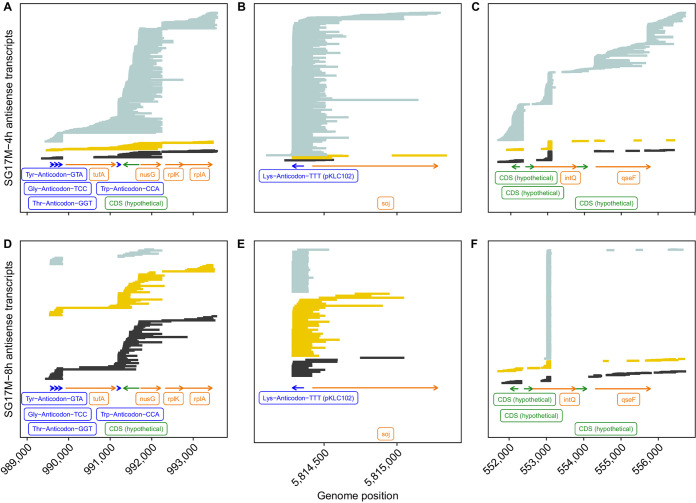
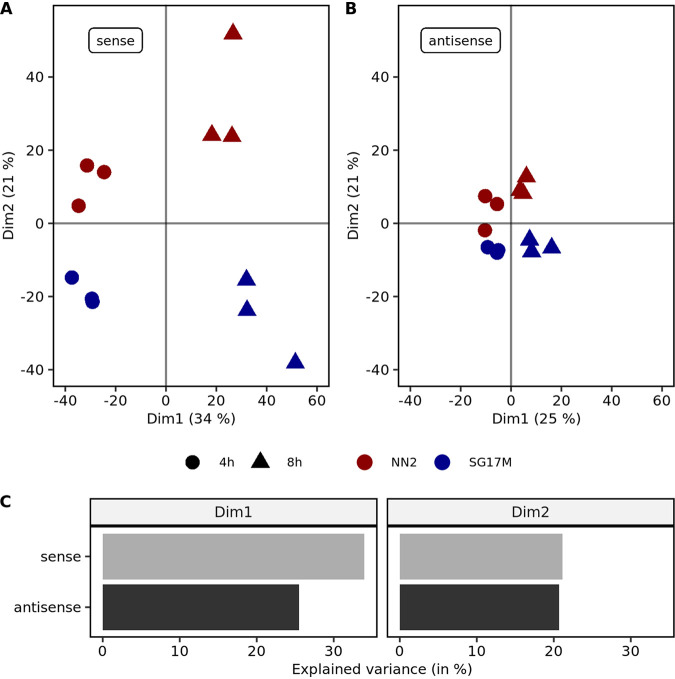
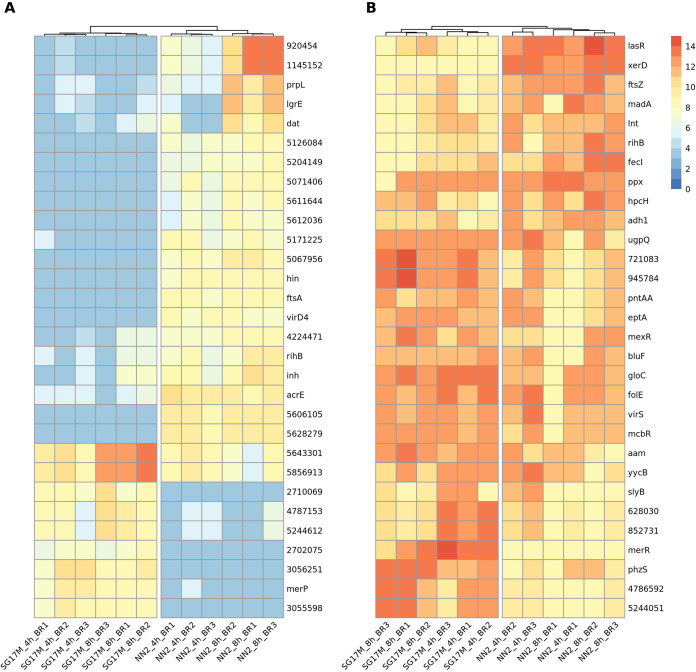
The transcriptomes of Pseudomonas aeruginosa clone C isolates NN2 and SG17M during the mid-exponential and early stationary phases of planktonic growth were evaluated by direct RNA sequencing on the nanopore platform and compared with established short-read cDNA sequencing on the Illumina platform. Fifty to ninety percent of the sense RNAs turned out to be rRNA molecules, followed by similar proportions of mRNA transcripts and noncoding RNAs. The two platforms detected similar proportions of uncharged tRNAs and 29 yet-undescribed antisense tRNAs. For example, the rarest arginine codon was paired with the most abundant tRNAArg, and the tRNAArg gene is missing for the most frequent arginine codon. More than 90% of the antisense RNA molecules were complementary to a coding sequence. The antisense RNAs were evenly distributed in the genomes. Direct RNA sequencing identified more than 4,000 distinct nonoverlapping antisense RNAs during exponential and stationary growth. Besides highly expressed small antisense RNAs less than 200 bases in size, a population of longer antisense RNAs was sequenced that covered a broad range (a few hundred to thousands of bases) and could be complementary to a contig of several genes. In summary, direct RNA sequencing identified yet-undescribed RNA molecules and an unexpected composition of the pools of tRNAs and sense and antisense RNAs. IMPORTANCE Genome-wide gene expression of bacteria is commonly studied by high-throughput sequencing of size-selected cDNA fragment libraries of reverse-transcribed RNA preparations. However, the depletion of rRNAs, enzymatic reverse transcription, and the fragmentation, size selection, and amplification during library preparation lead to inevitable losses of information about the initial composition of the RNA pool. We demonstrate that direct RNA sequencing on the Nanopore platform can overcome these limitations. Nanopore sequencing of total RNA yielded novel insights into the Pseudomonas aeruginosa transcriptome that-if replicated in other species-will change our view of the bacterial RNA world. The discovery of sense-antisense pairs of transfer-messenger RNA (tmRNA), tRNAs, and mRNAs indicates a further and unknown level of gene regulation in bacteria.
Keywords: Nanopore sequencing; Pseudomonas aeruginosa; antisense RNA; direct RNA sequencing; tRNA; transcriptome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Organisation for Economic Cooperation and Development. 1997. Consensus document on information used in the assessment of environmental applications involving Pseudomonas. https://www.oecd.org/env/ehs/biotrack/46815578.pdf.
-
- Freschi L, Vincent AT, Jeukens J, Emond-Rheault JG, Kukavica-Ibrulj I, Dupont MJ, Charette SJ, Boyle B, Levesque RC. 2019. The Pseudomonas aeruginosa pan-genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol Evol 11:109–120. 10.1093/gbe/evy259. - DOI - PMC - PubMed
-
- Hilker R, Munder A, Klockgether J, Losada PM, Chouvarine P, Cramer N, Davenport CF, Dethlefsen S, Fischer S, Peng H, Schönfelder T, Türk O, Wiehlmann L, Wölbeling F, Gulbins E, Goesmann A, Tümmler B. 2015. Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ Microbiol 17:29–46. 10.1111/1462-2920.12606. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
