

Chk1 Inhibition Potently Blocks STAT3 Tyrosine705 Phosphorylation, DNA-Binding Activity, and Activation of Downstream Targets in Human Multiple Myeloma Cells
- PMID: 34782371
- PMCID: PMC8898301
- DOI: 10.1158/1541-7786.MCR-21-0366
Chk1 Inhibition Potently Blocks STAT3 Tyrosine705 Phosphorylation, DNA-Binding Activity, and Activation of Downstream Targets in Human Multiple Myeloma Cells
Abstract
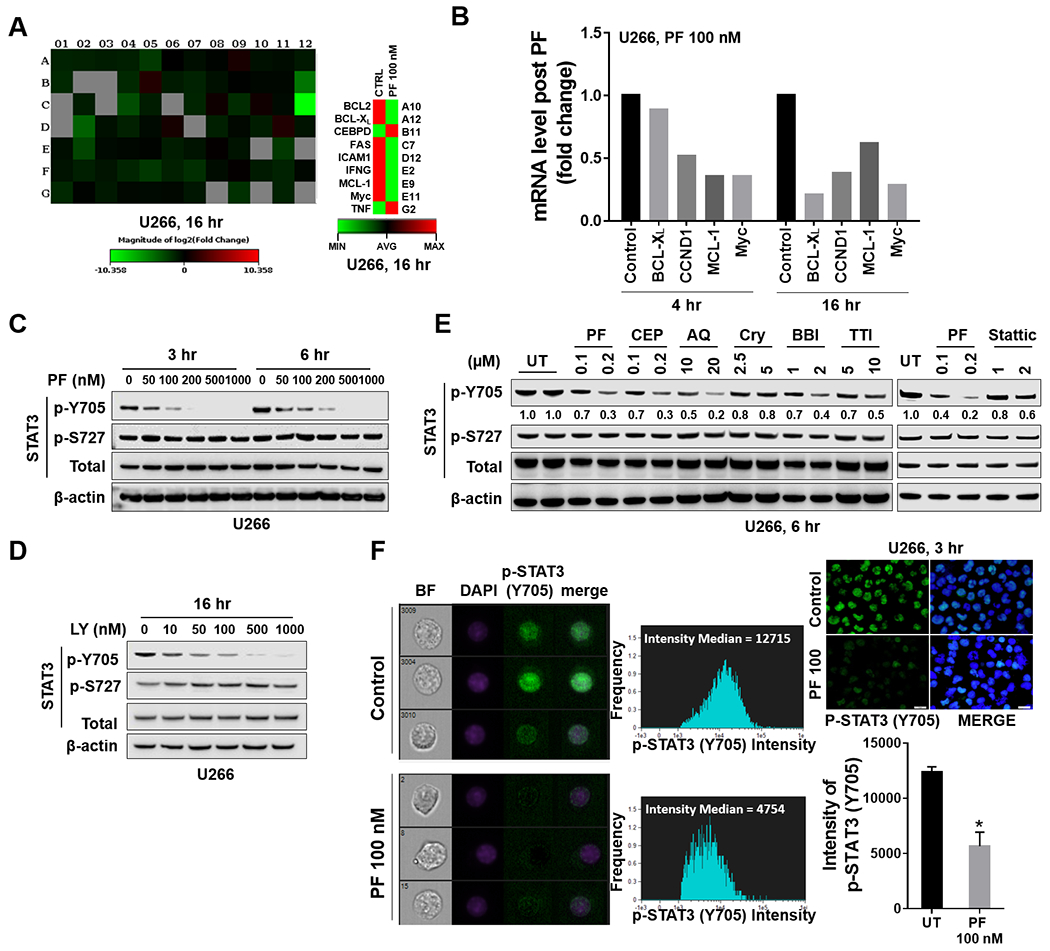
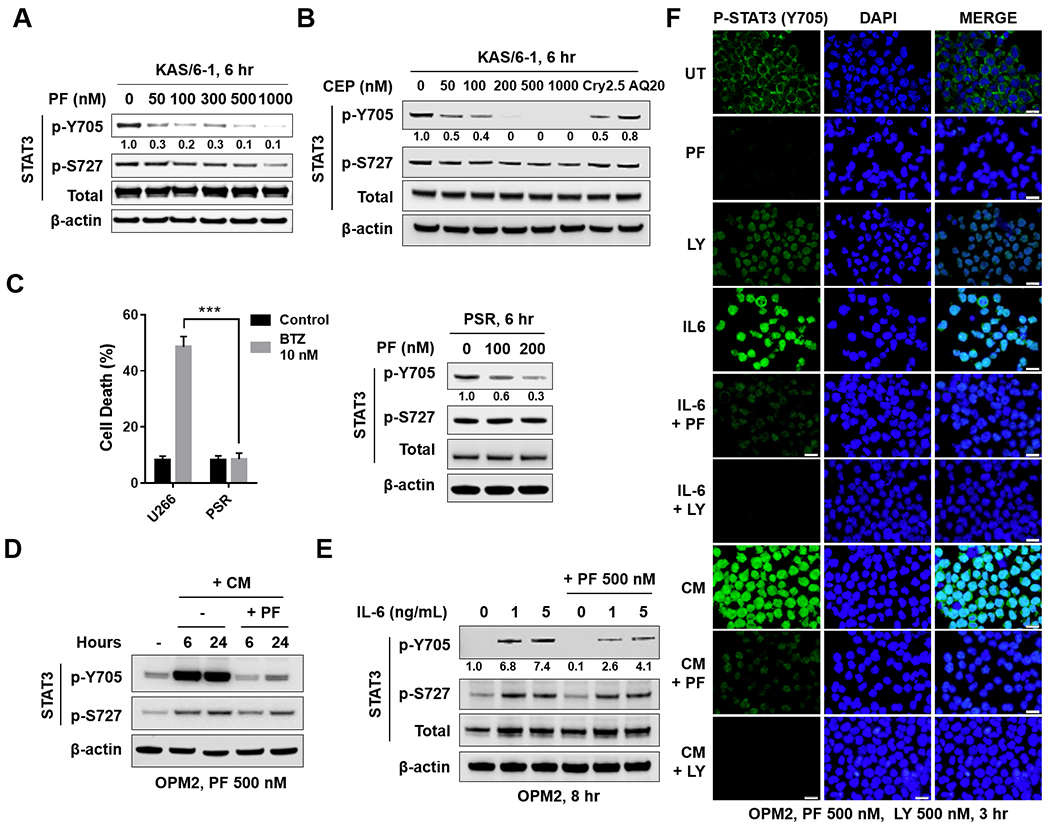
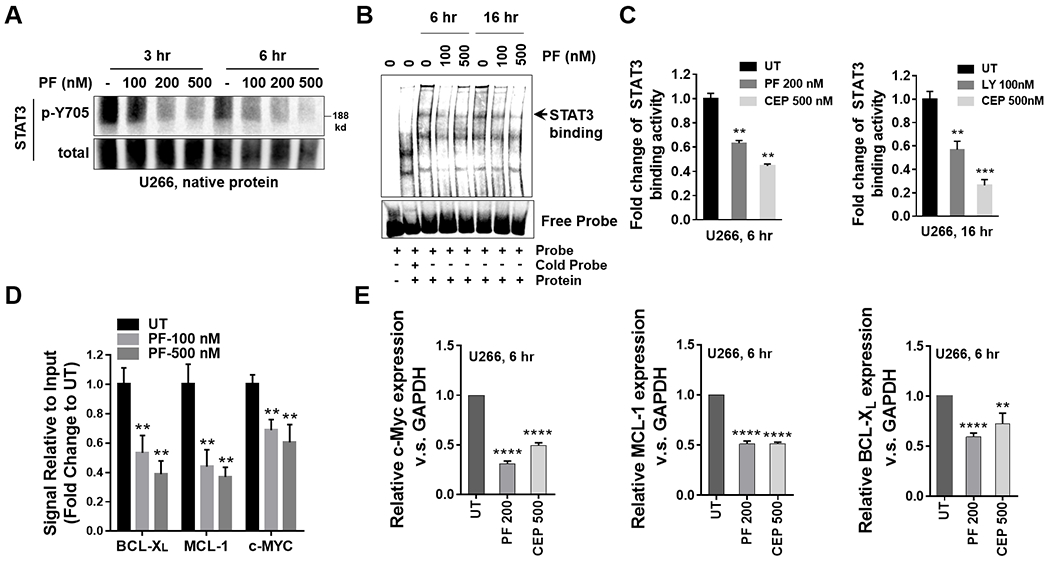
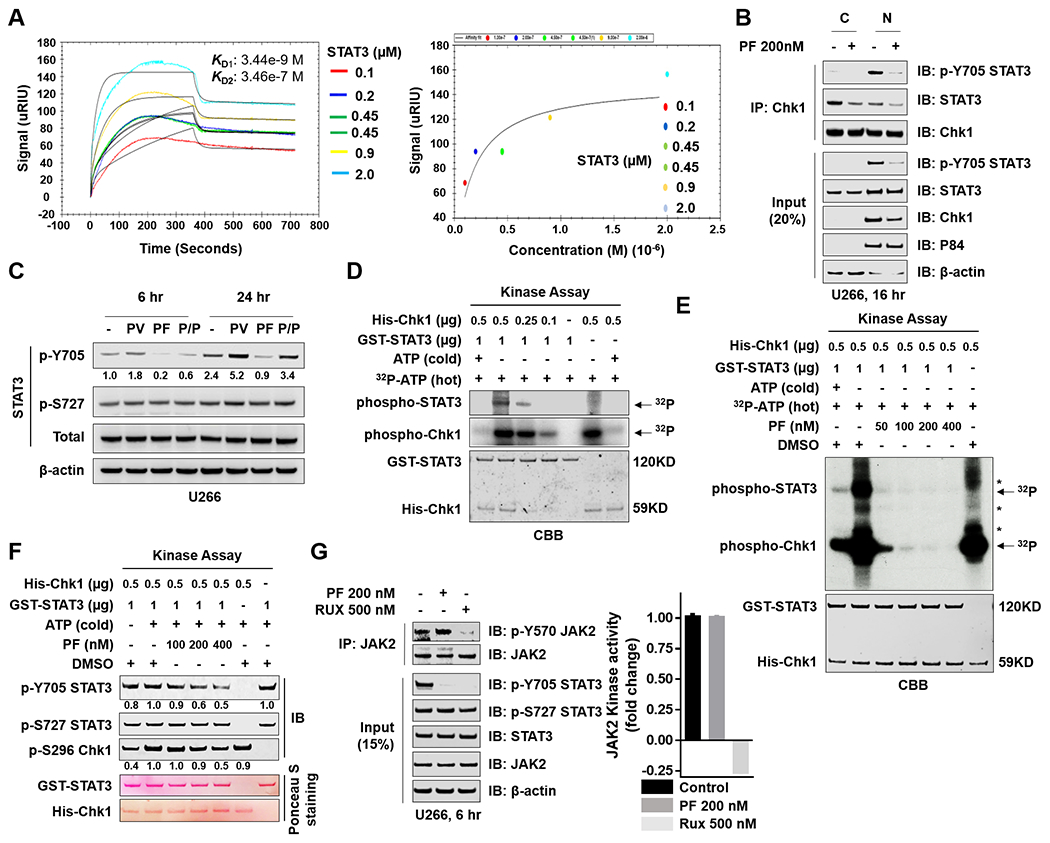
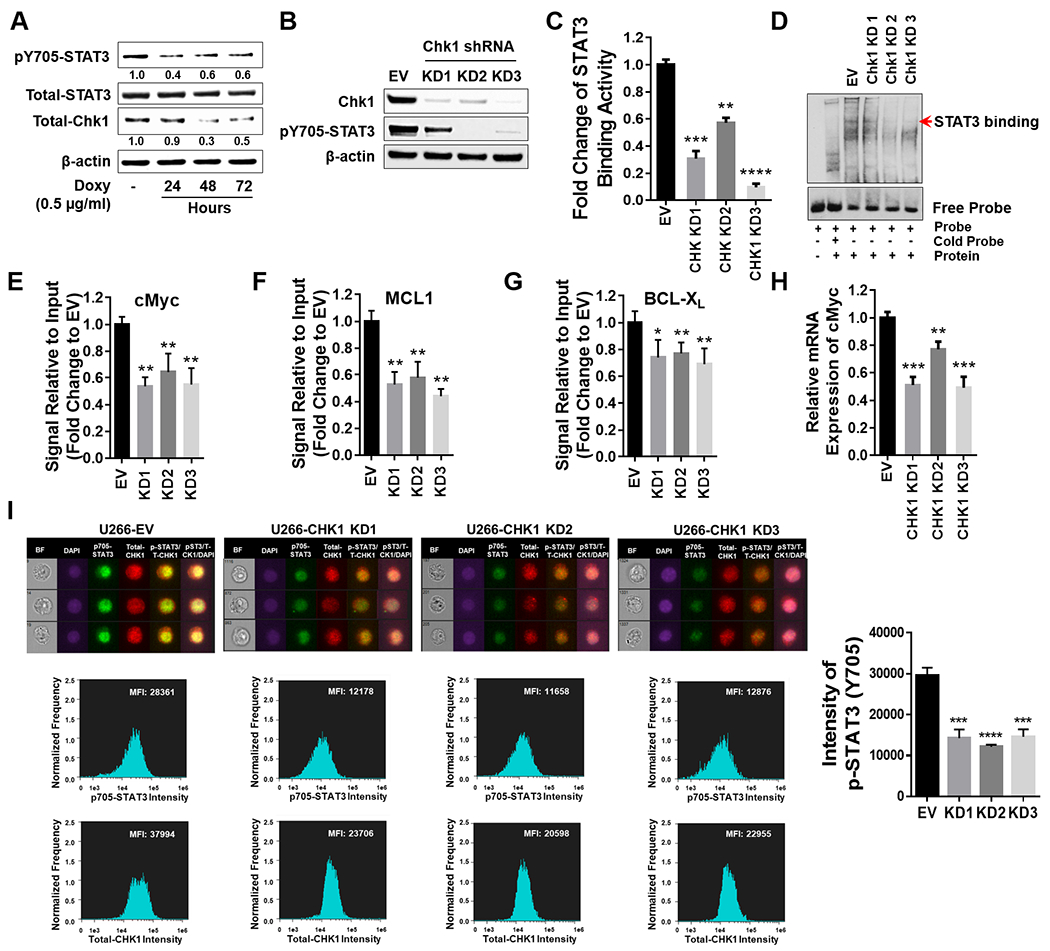
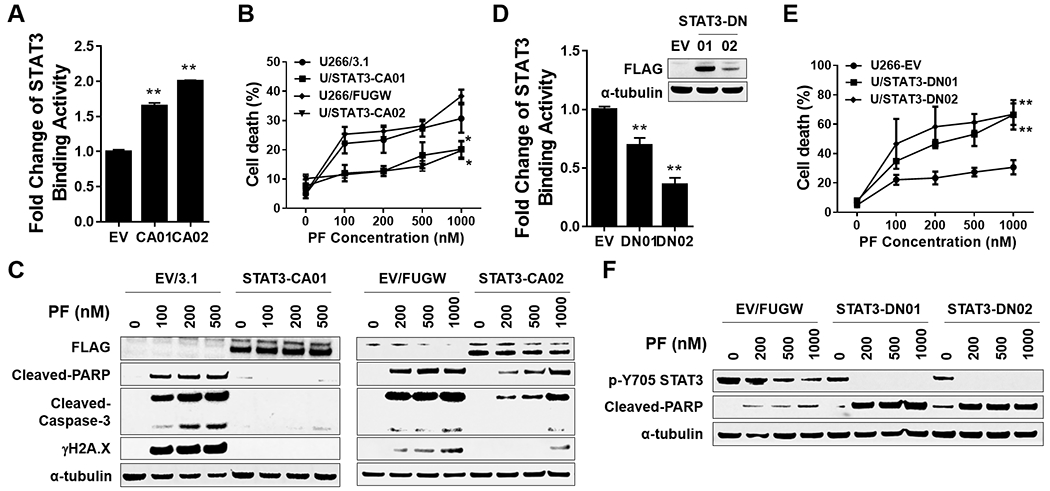
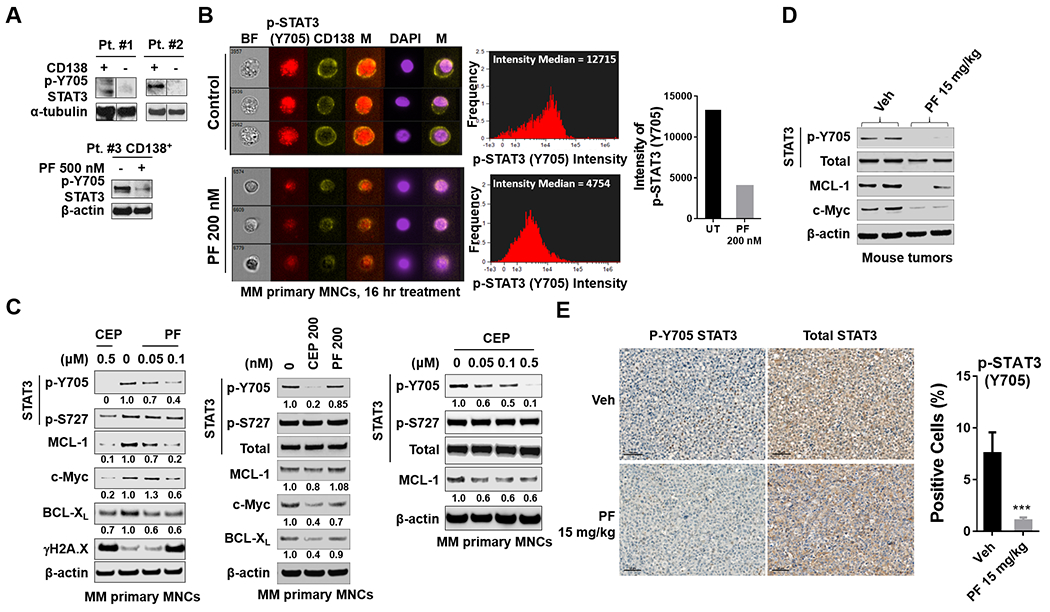
The relationship between the checkpoint kinase Chk1 and the STAT3 pathway was examined in multiple myeloma cells. Gene expression profiling of U266 cells exposed to low (nmol/L) Chk1 inhibitor [PF-477736 (PF)] concentrations revealed STAT3 pathway-related gene downregulation (e.g., BCL-XL, MCL-1, c-Myc), findings confirmed by RT-PCR. This was associated with marked inhibition of STAT3 Tyr705 (but not Ser727) phosphorylation, dimerization, nuclear localization, DNA binding, STAT3 promoter activity by chromatin immunoprecipitation assay, and downregulation of STAT-3-dependent proteins. Similar findings were obtained in other multiple myeloma cells and with alternative Chk1 inhibitors (e.g., prexasertib, CEP3891). While PF did not reduce GP130 expression or modify SOCS or PRL-3 phosphorylation, the phosphatase inhibitor pervanadate antagonized PF-mediated Tyr705 dephosphorylation. Significantly, PF attenuated Chk1-mediated STAT3 phosphorylation in in vitro assays. Surface plasmon resonance analysis suggested Chk1/STAT3 interactions and PF reduced Chk1/STAT3 co-immunoprecipitation. Chk1 CRISPR knockout or short hairpin RNA knockdown cells also displayed STAT3 inactivation and STAT3-dependent protein downregulation. Constitutively active STAT3 diminished PF-mediated STAT3 inactivation and downregulate STAT3-dependent proteins while significantly reducing PF-induced DNA damage (γH2A.X formation) and apoptosis. Exposure of cells with low basal phospho-STAT3 expression to IL6 or human stromal cell conditioned medium activated STAT3, an event attenuated by Chk1 inhibitors. PF also inactivated STAT3 in primary human CD138+ multiple myeloma cells and tumors extracted from an NSG multiple myeloma xenograft model while inhibiting tumor growth.
Implications: These findings identify a heretofore unrecognized link between the Chk1 and STAT3 pathways and suggest that Chk1 pathway inhibitors warrant attention as novel and potent candidate STAT3 antagonists in myeloma.
©2021 American Association for Cancer Research.
Conflict of interest statement
Figures
Similar articles
-
Combined MEK1/2 and ATR inhibition promotes myeloma cell death through a STAT3-dependent mechanism in vitro and in vivo.Br J Haematol. 2024 Dec;205(6):2338-2348. doi: 10.1111/bjh.19796. Epub 2024 Oct 8. Br J Haematol. 2024. PMID: 39379134
-
Non-canonical role for the ataxia-telangiectasia-Rad3 pathway in STAT3 activation in human multiple myeloma cells.Cell Oncol (Dordr). 2023 Oct;46(5):1369-1380. doi: 10.1007/s13402-023-00817-6. Epub 2023 May 1. Cell Oncol (Dordr). 2023. PMID: 37126127 Free PMC article.
-
Disruption of Src function potentiates Chk1-inhibitor-induced apoptosis in human multiple myeloma cells in vitro and in vivo.Blood. 2011 Feb 10;117(6):1947-57. doi: 10.1182/blood-2010-06-291146. Epub 2010 Dec 10. Blood. 2011. PMID: 21148814 Free PMC article.
-
IL6 Promotes a STAT3-PRL3 Feedforward Loop via SHP2 Repression in Multiple Myeloma.Cancer Res. 2019 Sep 15;79(18):4679-4688. doi: 10.1158/0008-5472.CAN-19-0343. Epub 2019 Jul 23. Cancer Res. 2019. PMID: 31337650
-
Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells.Blood. 2008 Sep 15;112(6):2439-49. doi: 10.1182/blood-2008-05-159392. Epub 2008 Jul 9. Blood. 2008. PMID: 18614762 Free PMC article.
Cited by
-
Development and validation of a mitotic catastrophe-related genes prognostic model for breast cancer.PeerJ. 2024 Sep 20;12:e18075. doi: 10.7717/peerj.18075. eCollection 2024. PeerJ. 2024. PMID: 39314848 Free PMC article.
-
Combined MEK1/2 and ATR inhibition promotes myeloma cell death through a STAT3-dependent mechanism in vitro and in vivo.Br J Haematol. 2024 Dec;205(6):2338-2348. doi: 10.1111/bjh.19796. Epub 2024 Oct 8. Br J Haematol. 2024. PMID: 39379134
-
Non-canonical role for the ataxia-telangiectasia-Rad3 pathway in STAT3 activation in human multiple myeloma cells.Cell Oncol (Dordr). 2023 Oct;46(5):1369-1380. doi: 10.1007/s13402-023-00817-6. Epub 2023 May 1. Cell Oncol (Dordr). 2023. PMID: 37126127 Free PMC article.
-
PNKP targeting engages the autophagic machinery through STING and STAT3 to potentiate ferroptosis and chemotherapy in TNBC.Redox Biol. 2025 Jul 22;86:103775. doi: 10.1016/j.redox.2025.103775. Online ahead of print. Redox Biol. 2025. PMID: 40743845 Free PMC article.
-
Src inhibition potentiates MCL-1 antagonist activity in acute myeloid leukemia.Signal Transduct Target Ther. 2025 Feb 10;10(1):50. doi: 10.1038/s41392-025-02125-x. Signal Transduct Target Ther. 2025. PMID: 39924517 Free PMC article.
References
-
- Siveen KS, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–154. - PubMed
-
- Aziz MH, Hafeez BB, Sand JM, et al. Protein kinase Cvarepsilon mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene. 2010;29(21):3100–3109. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
