

Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization
- PMID: 34782457
- PMCID: PMC8617501
- DOI: 10.1073/pnas.2100122118
Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization
Abstract
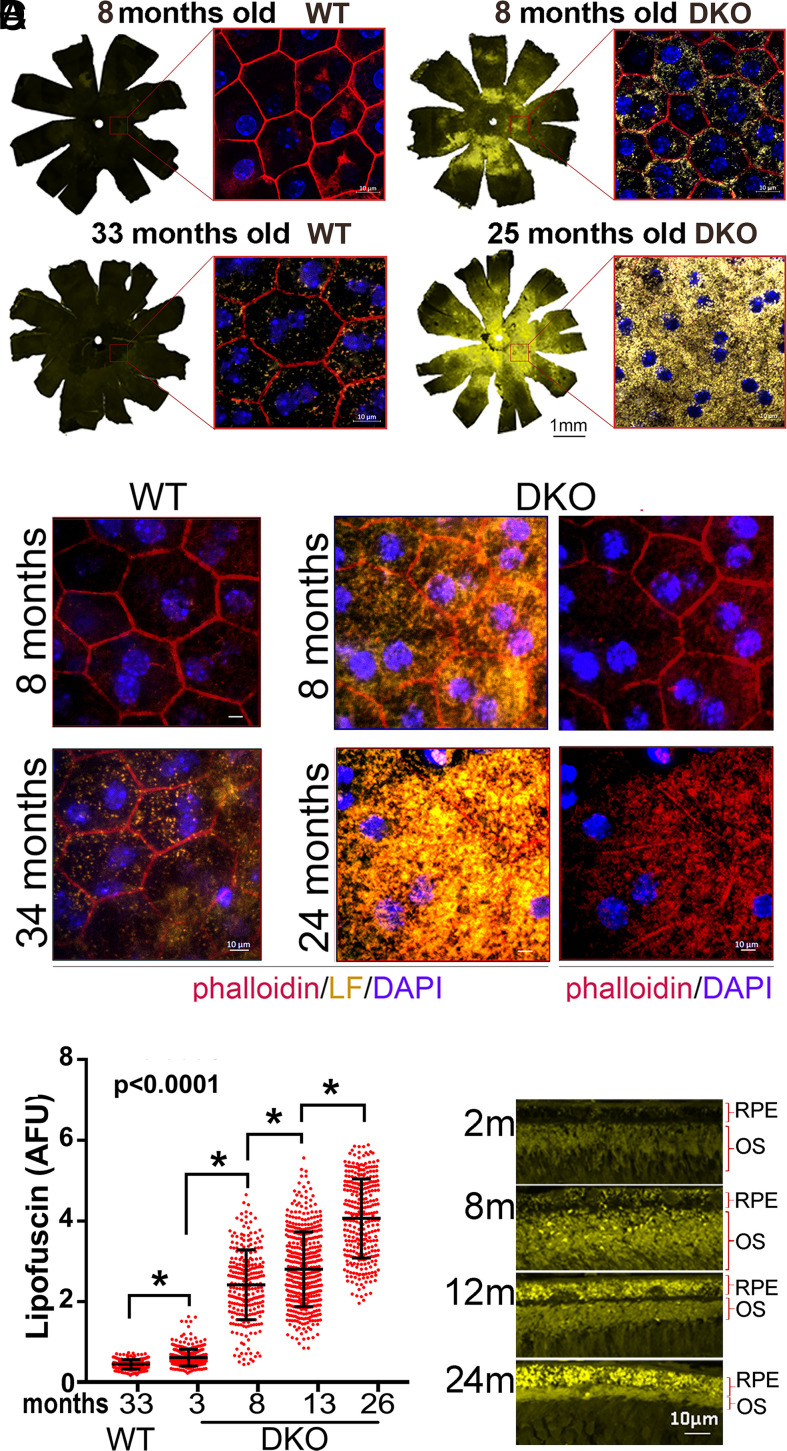
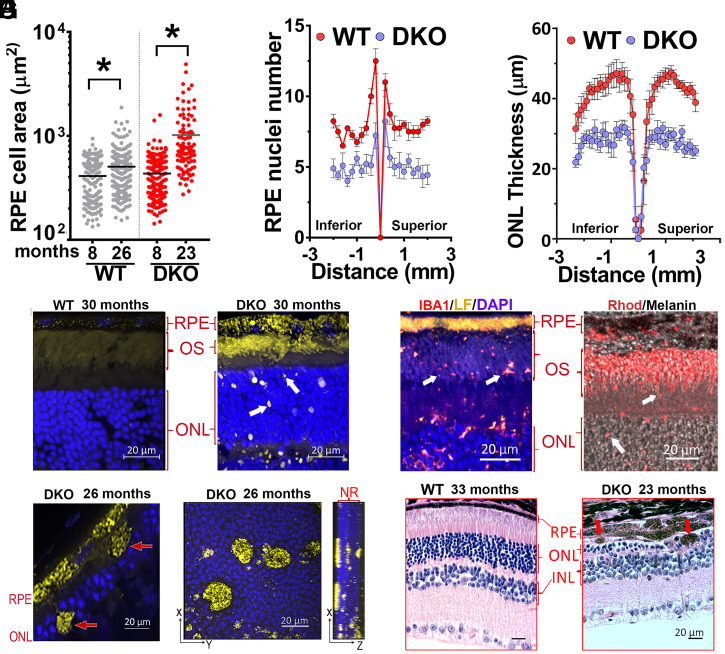
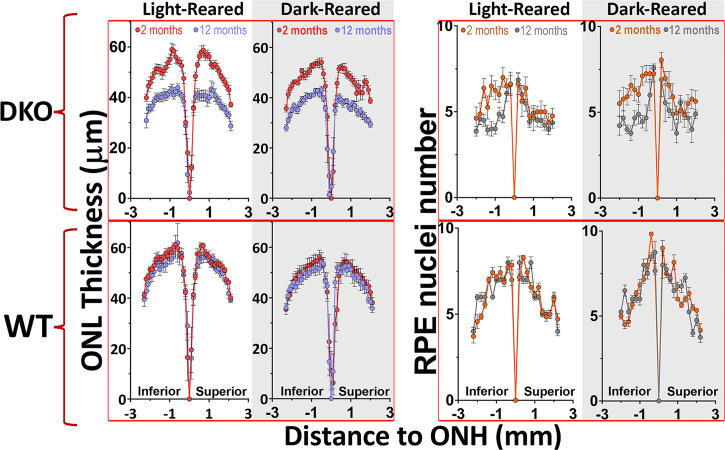
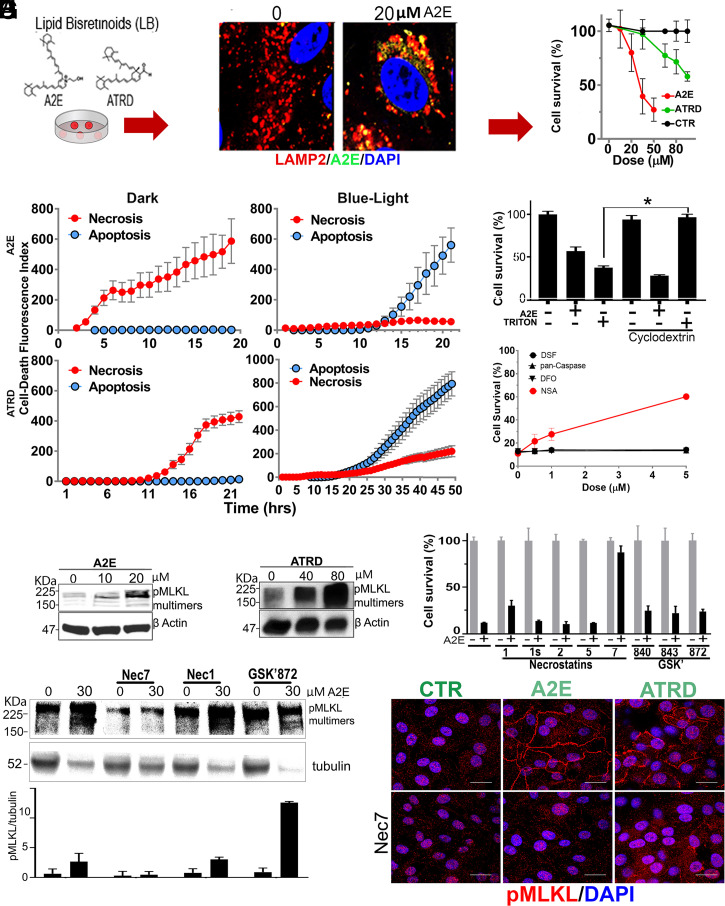
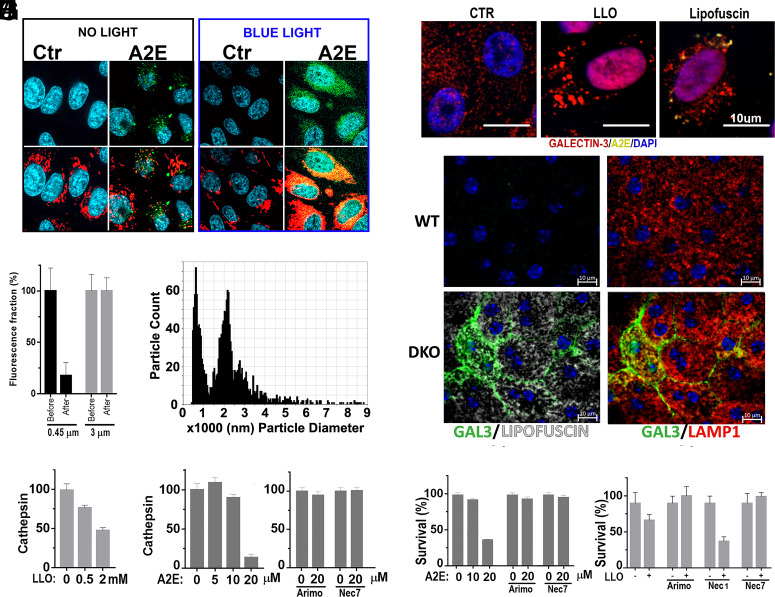
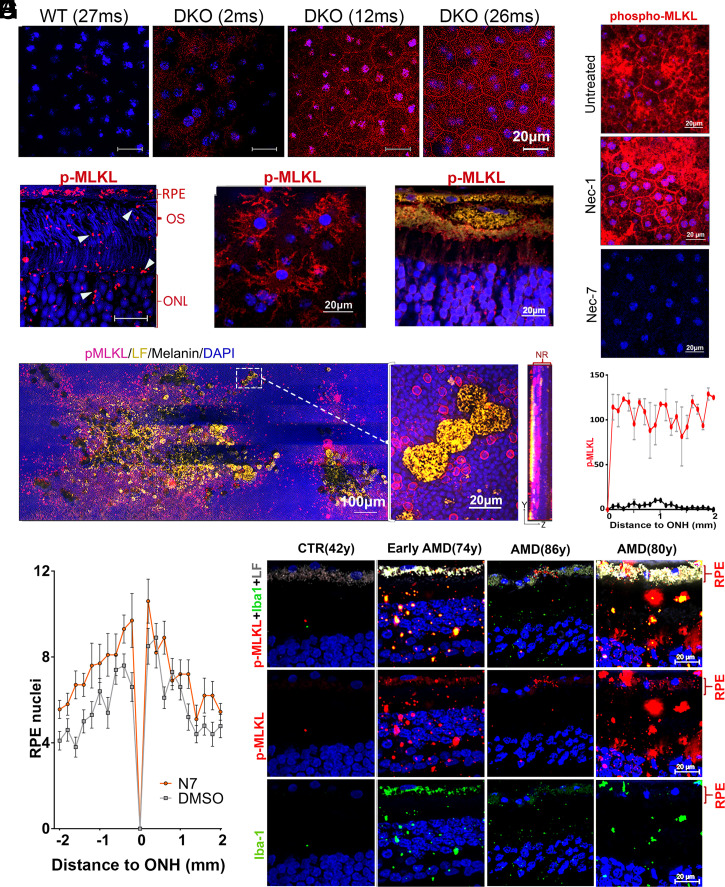
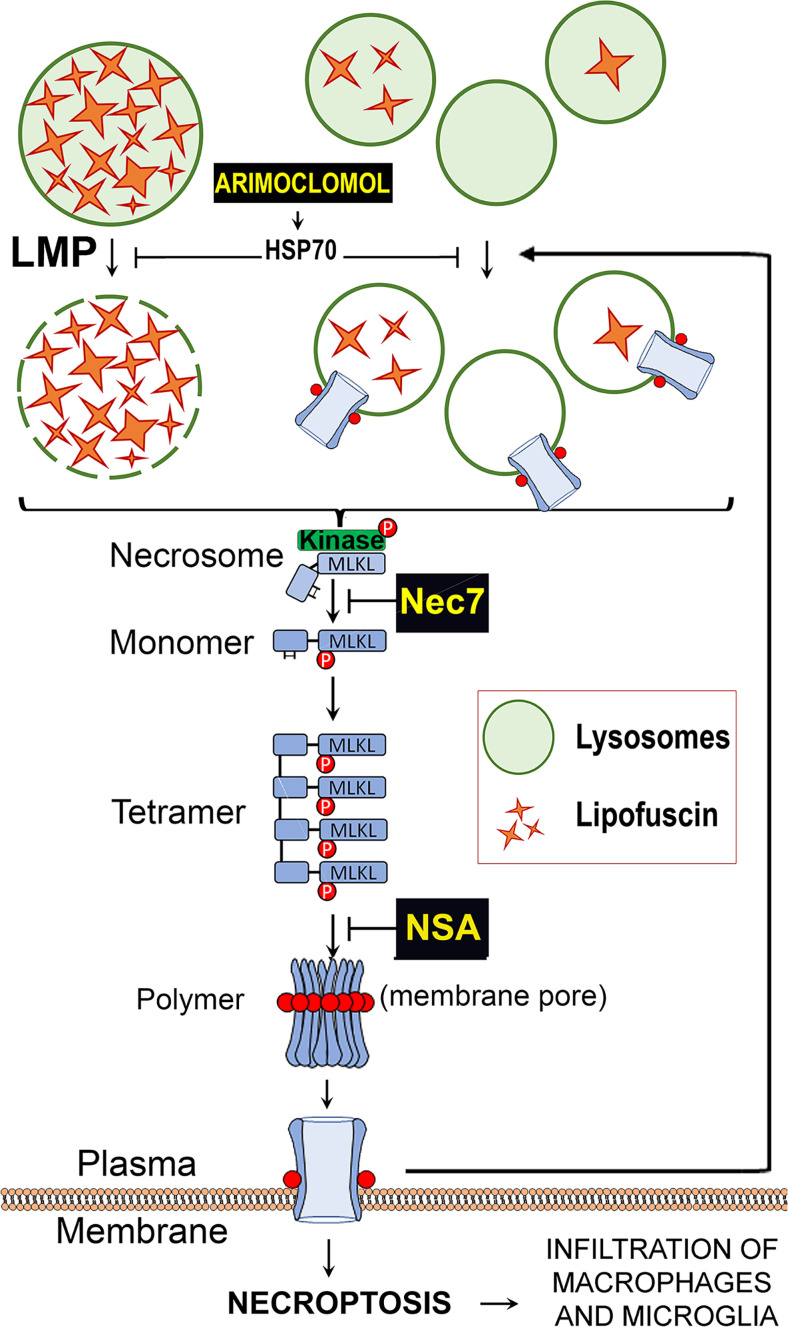
Lipofuscin granules enclose mixtures of cross-linked proteins and lipids in proportions that depend on the tissue analyzed. Retinal lipofuscin is unique in that it contains mostly lipids with very little proteins. However, retinal lipofuscin also presents biological and physicochemical characteristics indistinguishable from conventional granules, including indigestibility, tendency to cause lysosome swelling that results in rupture or defective functions, and ability to trigger NLRP3 inflammation, a symptom of low-level disruption of lysosomes. In addition, like conventional lipofuscins, it appears as an autofluorescent pigment, considered toxic waste, and a biomarker of aging. Ocular lipofuscin accumulates in the retinal pigment epithelium (RPE), whereby it interferes with the support of the neuroretina. RPE cell death is the primary cause of blindness in the most prevalent incurable genetic and age-related human disorders, Stargardt disease and age-related macular degeneration (AMD), respectively. Although retinal lipofuscin is directly linked to the cell death of the RPE in Stargardt, the extent to which it contributes to AMD is a matter of debate. Nonetheless, the number of AMD clinical trials that target lipofuscin formation speaks for the potential relevance for AMD as well. Here, we show that retinal lipofuscin triggers an atypical necroptotic cascade, amenable to pharmacological intervention. This pathway is distinct from canonic necroptosis and is instead dependent on the destabilization of lysosomes. We also provide evidence that necroptosis is activated in aged human retinas with AMD. Overall, this cytotoxicity mechanism may offer therapeutic targets and markers for genetic and age-related diseases associated with lipofuscin buildups.
Keywords: LMP; Lipofuscin; aging; lipid-bisretinoids; necroptosis.
Conflict of interest statement
Competing interest statement: A patent has been filed for targeting the LMP/necroptosis cascade.
Figures
Similar articles
-
Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration.Proc Natl Acad Sci U S A. 2018 Nov 20;115(47):E11120-E11127. doi: 10.1073/pnas.1802519115. Epub 2018 Nov 5. Proc Natl Acad Sci U S A. 2018. PMID: 30397118 Free PMC article.
-
Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage.J Mol Med (Berl). 2015 Aug;93(8):905-16. doi: 10.1007/s00109-015-1275-1. Epub 2015 Mar 18. J Mol Med (Berl). 2015. PMID: 25783493 Free PMC article.
-
Melatonin protects RPE cells from necroptosis and NLRP3 activation via promoting SERCA2-related intracellular Ca2+ homeostasis.Phytomedicine. 2024 Dec;135:156088. doi: 10.1016/j.phymed.2024.156088. Epub 2024 Sep 22. Phytomedicine. 2024. PMID: 39341129
-
Structures and biogenetic analysis of lipofuscin bis-retinoids.J Zhejiang Univ Sci B. 2013 Sep;14(9):763-73. doi: 10.1631/jzus.B1300051. J Zhejiang Univ Sci B. 2013. PMID: 24009196 Free PMC article. Review.
-
Retina and RPE lipid profile changes linked with ABCA4 associated Stargardt's maculopathy.Pharmacol Ther. 2023 Sep;249:108482. doi: 10.1016/j.pharmthera.2023.108482. Epub 2023 Jun 27. Pharmacol Ther. 2023. PMID: 37385300 Free PMC article. Review.
Cited by
-
Lipid Droplet Accumulation Promotes RPE Dysfunction.Int J Mol Sci. 2022 Feb 4;23(3):1790. doi: 10.3390/ijms23031790. Int J Mol Sci. 2022. PMID: 35163712 Free PMC article.
-
Structure, Function, and Molecular Landscapes of the Aging Retina.Annu Rev Vis Sci. 2023 Sep 15;9:177-199. doi: 10.1146/annurev-vision-112122-020950. Epub 2023 May 17. Annu Rev Vis Sci. 2023. PMID: 37196423 Free PMC article. Review.
-
Exposure of A2E to blue light promotes ferroptosis in the retinal pigment epithelium.Cell Mol Biol Lett. 2025 Feb 21;30(1):22. doi: 10.1186/s11658-025-00700-2. Cell Mol Biol Lett. 2025. PMID: 39984833 Free PMC article.
-
Age-Related Lysosomal Dysfunctions.Cells. 2022 Jun 20;11(12):1977. doi: 10.3390/cells11121977. Cells. 2022. PMID: 35741106 Free PMC article. Review.
-
AIM2 activation mediated by RIPK1/3-dependent mitochondrial DNA release drives Aβ1-40-Induced retinal pigment epithelium injury.Cell Commun Signal. 2025 Jun 21;23(1):301. doi: 10.1186/s12964-025-02294-w. Cell Commun Signal. 2025. PMID: 40544279 Free PMC article.
References
-
- Gómez-Sintes R., Ledesma M. D., Boya P., Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 32, 150–168 (2016). - PubMed
-
- Brunk U., Brun A., The effect of aging on lysosomal permeability in nerve cells of the central nervous system. An enzyme histochemical study in rat. Histochemie 30, 315–324 (1972). - PubMed
-
- Sparrow J. R., Cai B., Jang Y. P., Zhou J., Nakanishi K., A2E, a Fluorophore of RPE Lipofuscin, Can Destabilize Membrane in Advances in Experimental Medicine and Biology, 2005, pp. 63–68. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
