

Standardizing workflows in imaging transcriptomics with the abagen toolbox
- PMID: 34783653
- PMCID: PMC8660024
- DOI: 10.7554/eLife.72129
Standardizing workflows in imaging transcriptomics with the abagen toolbox
Abstract
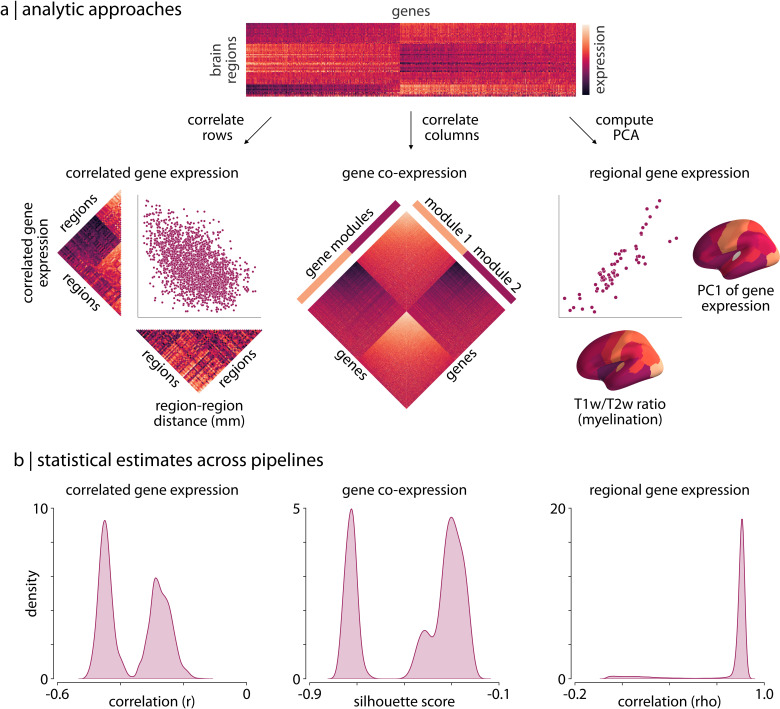
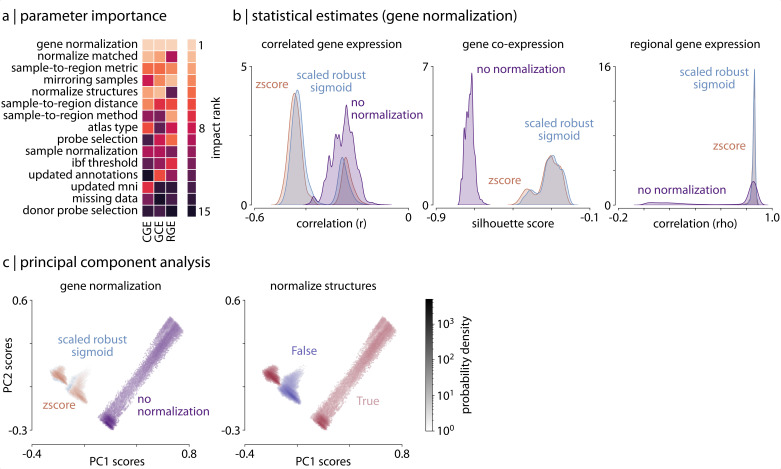
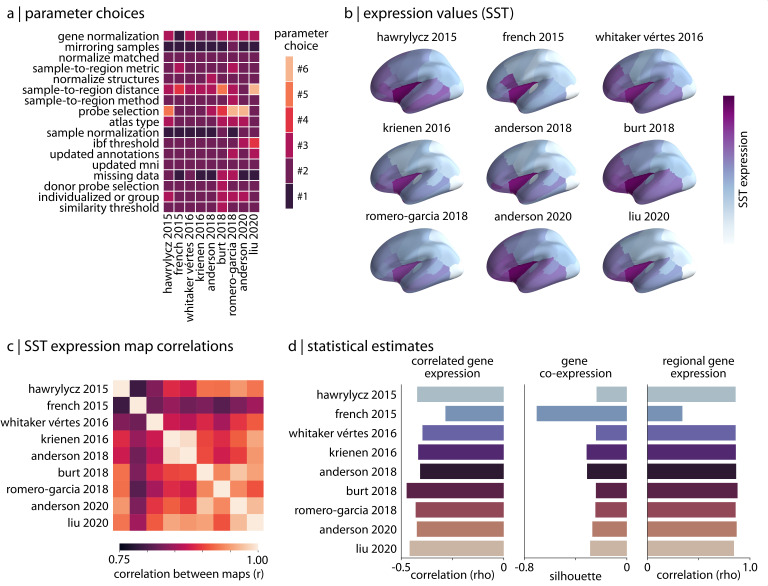
Gene expression fundamentally shapes the structural and functional architecture of the human brain. Open-access transcriptomic datasets like the Allen Human Brain Atlas provide an unprecedented ability to examine these mechanisms in vivo; however, a lack of standardization across research groups has given rise to myriad processing pipelines for using these data. Here, we develop the abagen toolbox, an open-access software package for working with transcriptomic data, and use it to examine how methodological variability influences the outcomes of research using the Allen Human Brain Atlas. Applying three prototypical analyses to the outputs of 750,000 unique processing pipelines, we find that choice of pipeline has a large impact on research findings, with parameters commonly varied in the literature influencing correlations between derived gene expression and other imaging phenotypes by as much as ρ ≥ 1.0. Our results further reveal an ordering of parameter importance, with processing steps that influence gene normalization yielding the greatest impact on downstream statistical inferences and conclusions. The presented work and the development of the abagen toolbox lay the foundation for more standardized and systematic research in imaging transcriptomics, and will help to advance future understanding of the influence of gene expression in the human brain.
Keywords: MRI; human; neuroimaging; neuroscience; processing variability; software; transcriptomics.
© 2021, Markello et al.
Conflict of interest statement
RM, AA, JP, BF, AF, BM No competing interests declared
Figures


References
-
- Allen Institute for Brain Science . Allen Institute Publications for Brain Science; 2013. https://help.brain-map.org/display/humanbrain/Documentation
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
