

Nipah virus W protein harnesses nuclear 14-3-3 to inhibit NF-κB-induced proinflammatory response
- PMID: 34785771
- PMCID: PMC8595879
- DOI: 10.1038/s42003-021-02797-5
Nipah virus W protein harnesses nuclear 14-3-3 to inhibit NF-κB-induced proinflammatory response
Abstract
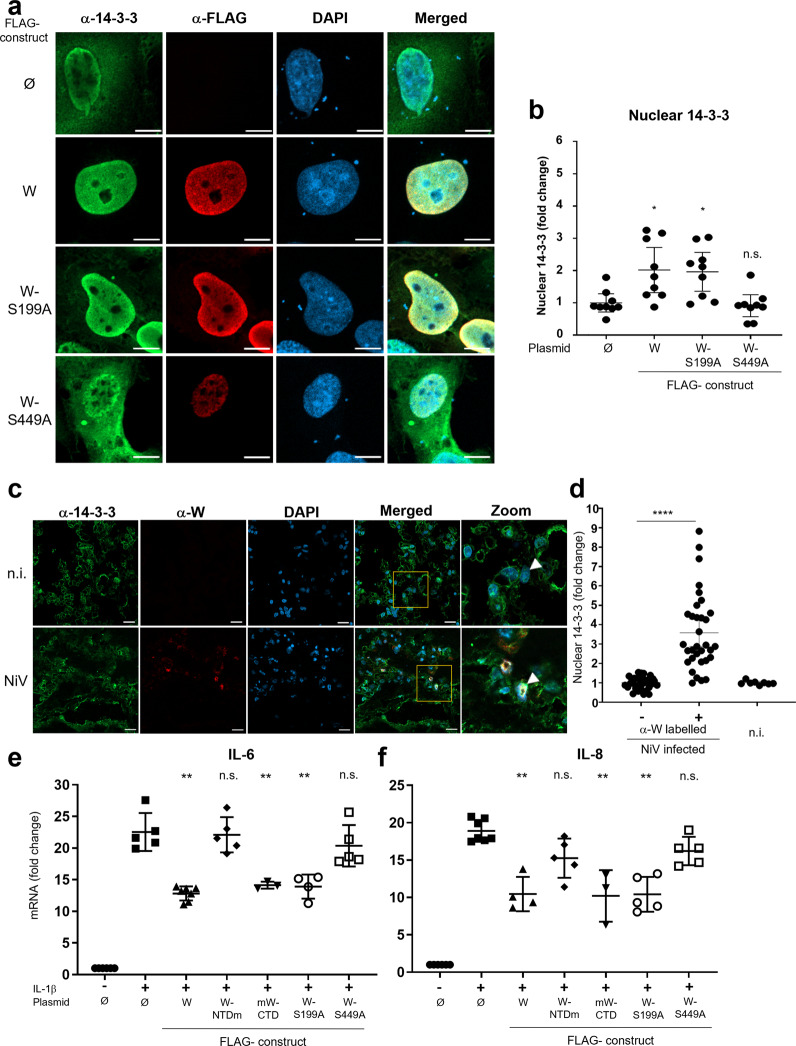
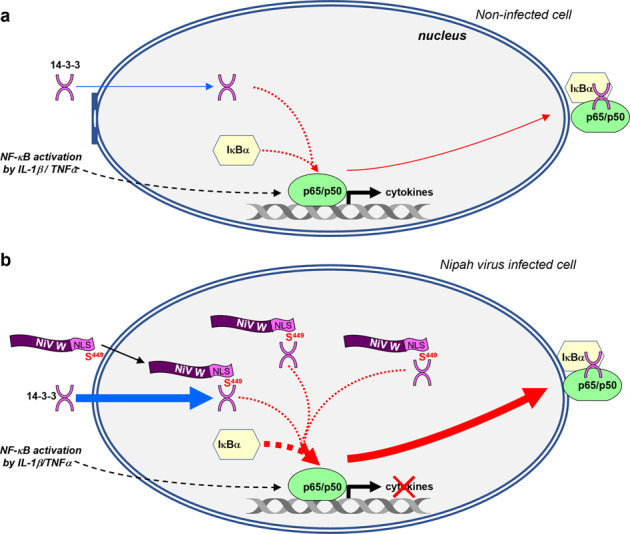
Nipah virus (NiV) is a highly pathogenic emerging bat-borne Henipavirus that has caused numerous outbreaks with public health concerns. It is able to inhibit the host innate immune response. Since the NF-κB pathway plays a crucial role in the innate antiviral response as a major transcriptional regulator of inflammation, we postulated its implication in the still poorly understood NiV immunopathogenesis. We report here that NiV inhibits the canonical NF-κB pathway via its nonstructural W protein. Translocation of the W protein into the nucleus causes nuclear accumulation of the cellular scaffold protein 14-3-3 in both African green monkey and human cells infected by NiV. Excess of 14-3-3 in the nucleus was associated with a reduction of NF-κB p65 subunit phosphorylation and of its nuclear accumulation. Importantly, W-S449A substitution impairs the binding of the W protein to 14-3-3 and the subsequent suppression of NF-κB signaling, thus restoring the production of proinflammatory cytokines. Our data suggest that the W protein increases the steady-state level of 14-3-3 in the nucleus and consequently enhances 14-3-3-mediated negative feedback on the NF-κB pathway. These findings provide a mechanistic model of W-mediated disruption of the host inflammatory response, which could contribute to the high severity of NiV infection.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
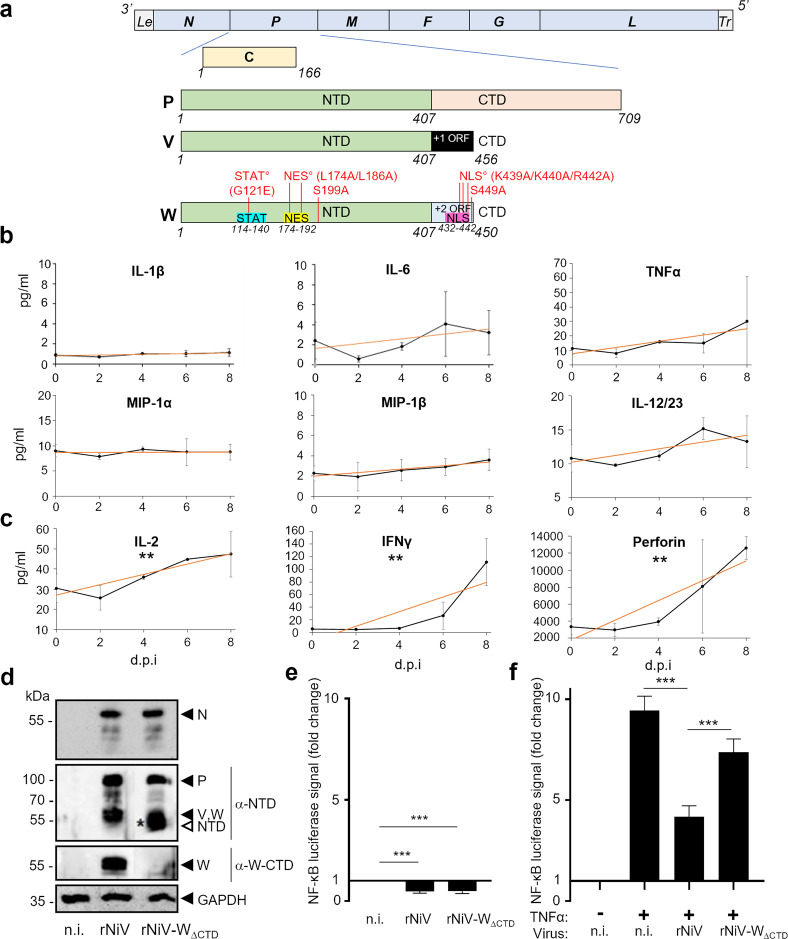
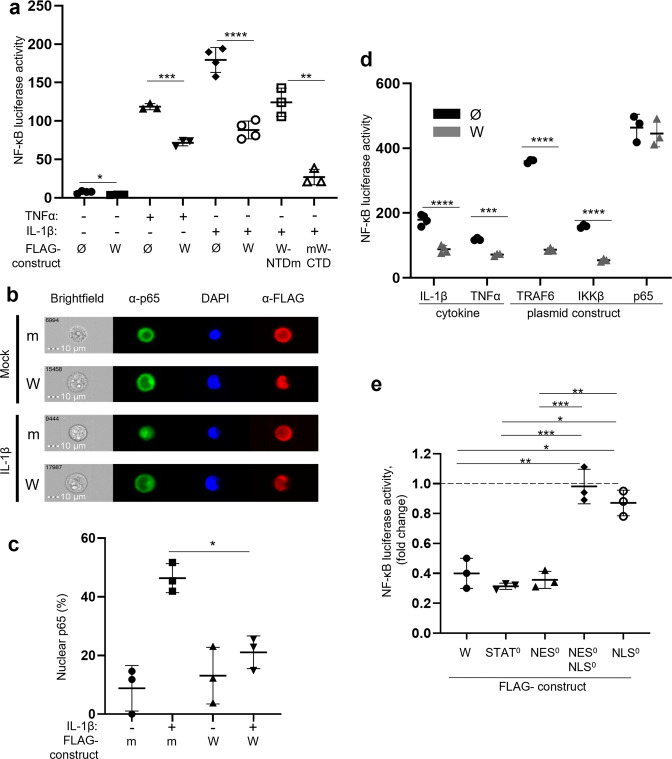


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
