

Automated proteomic sample preparation: The key component for high throughput and quantitative mass spectrometry analysis
- PMID: 34786750
- PMCID: PMC10339360
- DOI: 10.1002/mas.21750
Automated proteomic sample preparation: The key component for high throughput and quantitative mass spectrometry analysis
Abstract
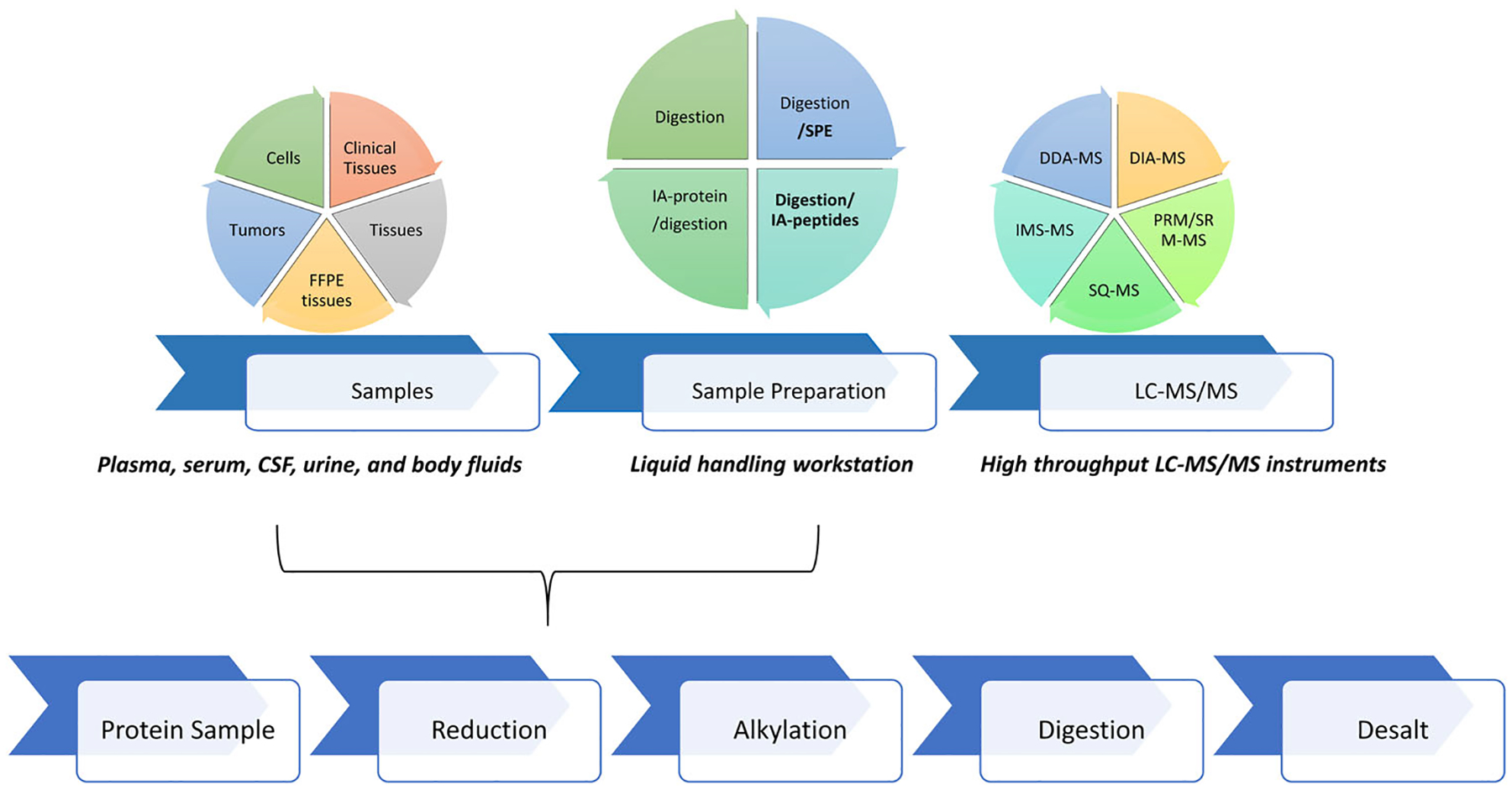
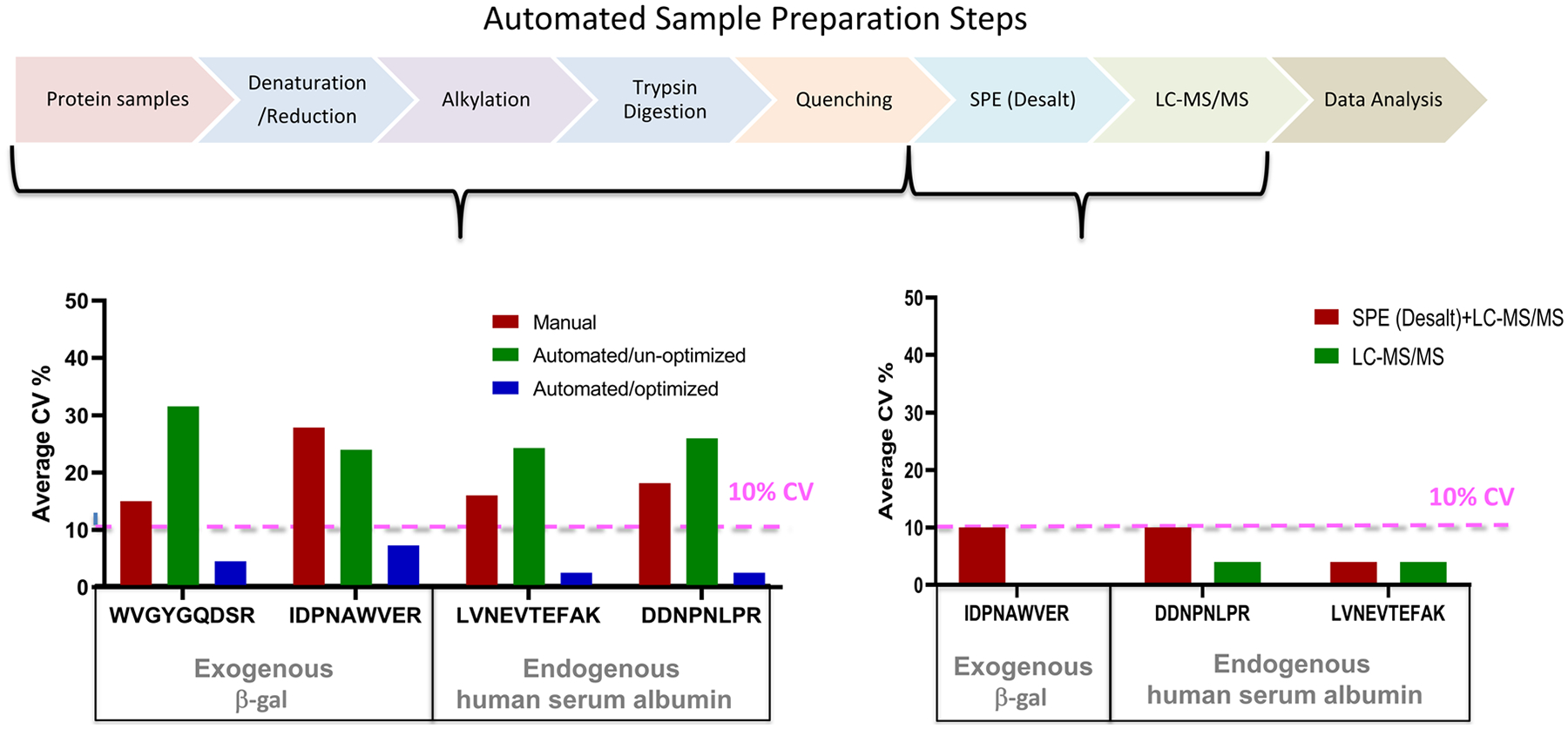
Sample preparation for mass spectrometry-based proteomics has many tedious and time-consuming steps that can introduce analytical errors. In particular, the steps around the proteolytic digestion of protein samples are prone to inconsistency. One route for reliable sample processing is the development and optimization of a workflow utilizing an automated liquid handling workstation. Diligent assessment of the sample type, protocol design, reagents, and incubation conditions can significantly improve the speed and consistency of preparation. When combining robust liquid chromatography-mass spectrometry with either discovery or targeted methods, automated sample preparation facilitates increased throughput and reproducible quantitation of biomarker candidates. These improvements in analysis are also essential to process the large patient cohorts necessary to validate a candidate biomarker for potential clinical use. This article reviews the steps in the workflow, optimization strategies, and known applications in clinical, pharmaceutical, and research fields that demonstrate the broad utility for improved automation of sample preparation in the proteomic field.
Keywords: automation; high throughput; optimization; proteomics; quantitative mass spectrometry; robot; sample preparation.
© 2021 John Wiley & Sons Ltd.
Figures
Similar articles
-
Highly Reproducible Automated Proteomics Sample Preparation Workflow for Quantitative Mass Spectrometry.J Proteome Res. 2018 Jan 5;17(1):420-428. doi: 10.1021/acs.jproteome.7b00623. Epub 2017 Nov 10. J Proteome Res. 2018. PMID: 29083196 Free PMC article.
-
High throughput and accurate serum proteome profiling by integrated sample preparation technology and single-run data independent mass spectrometry analysis.J Proteomics. 2018 Mar 1;174:9-16. doi: 10.1016/j.jprot.2017.12.014. Epub 2017 Dec 24. J Proteomics. 2018. PMID: 29278786
-
Automated "Cells-To-Peptides" Sample Preparation Workflow for High-Throughput, Quantitative Proteomic Assays of Microbes.J Proteome Res. 2019 Oct 4;18(10):3752-3761. doi: 10.1021/acs.jproteome.9b00455. Epub 2019 Aug 30. J Proteome Res. 2019. PMID: 31436101
-
[Advances in high-throughput proteomic analysis].Se Pu. 2021 Feb;39(2):112-117. doi: 10.3724/SP.J.1123.2020.08023. Se Pu. 2021. PMID: 34227342 Free PMC article. Review. Chinese.
-
A simplified optimization approach for sample preparation workflow in LC-MS-based quantitative proteomic analysis: Biological samples to peptides.Arch Pharm (Weinheim). 2025 Mar;358(3):e2400911. doi: 10.1002/ardp.202400911. Arch Pharm (Weinheim). 2025. PMID: 40038882 Review.
Cited by
-
Establishing Quality Control Metrics for Large-Scale Plasma Proteomic Sample Preparation.ACS Meas Sci Au. 2024 Apr 29;4(4):442-451. doi: 10.1021/acsmeasuresciau.3c00070. eCollection 2024 Aug 21. ACS Meas Sci Au. 2024. PMID: 39184360 Free PMC article.
-
The Future of Proteomics is Up in the Air: Can Ion Mobility Replace Liquid Chromatography for High Throughput Proteomics?J Proteome Res. 2024 Jun 7;23(6):1871-1882. doi: 10.1021/acs.jproteome.4c00248. Epub 2024 May 7. J Proteome Res. 2024. PMID: 38713528 Free PMC article. Review.
-
Parallelization with Dual-Trap Single-Column Configuration Maximizes Throughput of Proteomic Analysis.Anal Chem. 2022 Sep 13;94(36):12452-12460. doi: 10.1021/acs.analchem.2c02609. Epub 2022 Aug 31. Anal Chem. 2022. PMID: 36044770 Free PMC article.
-
Evaluating First-Pass, High Protein Capacity Desalting Techniques For Phosphoproteomics Applications.bioRxiv [Preprint]. 2025 Jun 3:2025.06.03.657744. doi: 10.1101/2025.06.03.657744. bioRxiv. 2025. PMID: 40501759 Free PMC article. Preprint.
-
Plasma/Serum Proteomics based on Mass Spectrometry.Protein Pept Lett. 2024;31(3):192-208. doi: 10.2174/0109298665286952240212053723. Protein Pept Lett. 2024. PMID: 38869039 Free PMC article. Review.
References
-
- An B, Zhang M, Pu J, Qu Y, Shen S, Zhou S, Ferrari L, Vazvaei F, and Qu J. 2020. ‘Toward accurate and robust liquid chromatography‐mass spectrometry‐based quantification of antibody biotherapeutics in tissues’, Anal Chem, 92: 15152–61. - PubMed
-
- Aslam B, Basit M, Nisar MA, Khurshid M, and Rasool MH. 2017. ‘Proteomics: Technologies and Their Applications’, J Chromatogr Sci, 55: 182–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
