

SARS-CoV-2 Variants and Their Relevant Mutational Profiles: Update Summer 2021
- PMID: 34787497
- PMCID: PMC8597642
- DOI: 10.1128/Spectrum.01096-21
SARS-CoV-2 Variants and Their Relevant Mutational Profiles: Update Summer 2021
Abstract
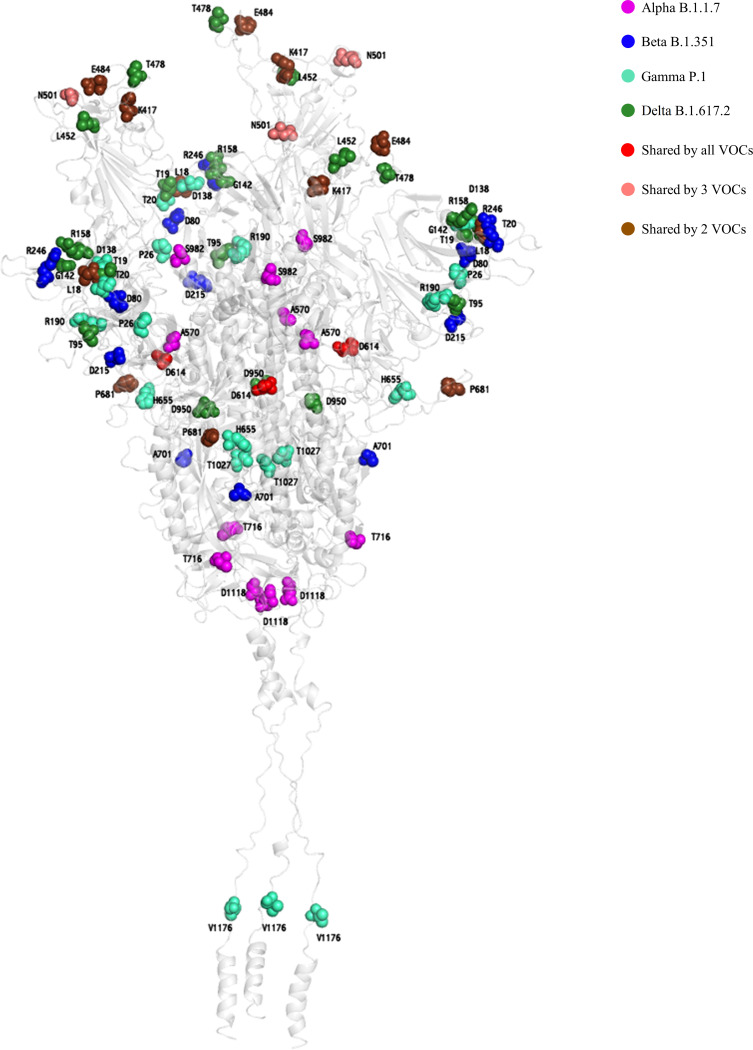
Since the beginning of the coronavirus disease 2019 (COVID-19) pandemic caused by it, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been undergoing a genetic diversification leading to the emergence of new variants. Nevertheless, a clear definition of the genetic signatures underlying the circulating variants is still missing. Here, we provide a comprehensive insight into mutational profiles characterizing each SARS-CoV-2 variant, focusing on spike mutations known to modulate viral infectivity and/or antigenicity. We focused on variants and on specific relevant mutations reported by GISAID, Nextstrain, Outbreak.info, Pango, and Stanford database websites that were associated with any clinical/diagnostic impact, according to published manuscripts. Furthermore, 1,223,338 full-length high-quality SARS-CoV-2 genome sequences were retrieved from GISAID and used to accurately define the specific mutational patterns in each variant. Finally, mutations were mapped on the three-dimensional structure of the SARS-CoV-2 spike protein to assess their localization in the different spike domains. Overall, this review sheds light and assists in defining the genetic signatures characterizing the currently circulating variants and their clinical relevance. IMPORTANCE Since the emergence of SARS-CoV-2, several recurrent mutations, particularly in the spike protein, arose during human-to-human transmission or spillover events between humans and animals, generating distinct worrisome variants of concern (VOCs) or of interest (VOIs), designated as such due to their clinical and diagnostic impacts. Characterizing these variants and their related mutations is important in tracking SAR-CoV-2 evolution and understanding the efficacy of vaccines and therapeutics based on monoclonal antibodies, convalescent-phase sera, and direct antivirals. Our study provides a comprehensive survey of the mutational profiles characterizing the important SARS-CoV-2 variants, focusing on spike mutations and highlighting other protein mutations.
Keywords: COVID-19; SARS-CoV-2; emerging variants; mutations; pandemic; variants.
Conflict of interest statement
Robert Shafer has received grant funding from Janssen Pharmaceuticals, Vela Diagnostics, and Insilixa and honoraria from Gilead Sciences and GlaxoSmithKline (GSK).
Figures


References
-
- Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, Masciovecchio C, Angeletti S, Ciccozzi M, Gallo RC, Zella D, Ippodrino R. 2020. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med 18:179. doi:10.1186/s12967-020-02344-6. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
