

Human B cell lineages associated with germinal centers following influenza vaccination are measurably evolving
- PMID: 34787567
- PMCID: PMC8741214
- DOI: 10.7554/eLife.70873
Human B cell lineages associated with germinal centers following influenza vaccination are measurably evolving
Abstract
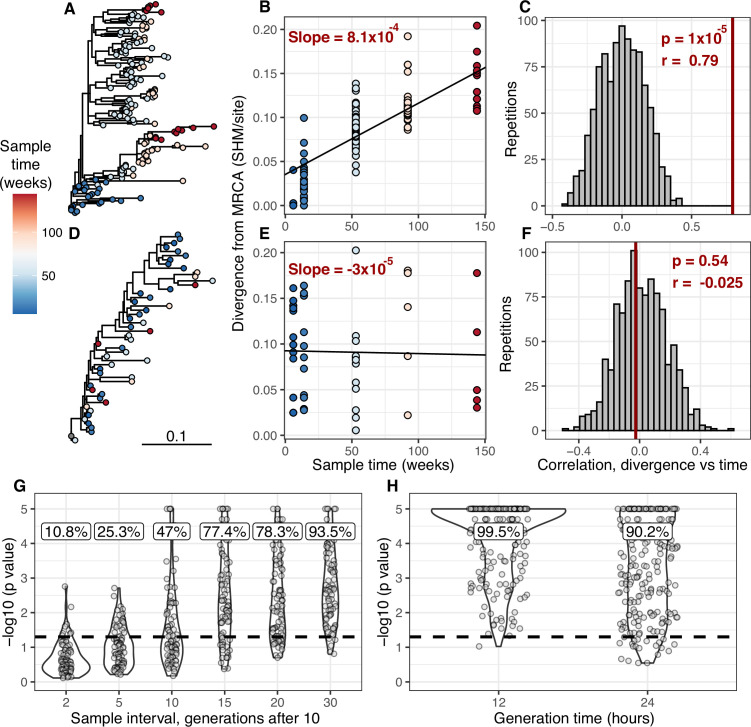
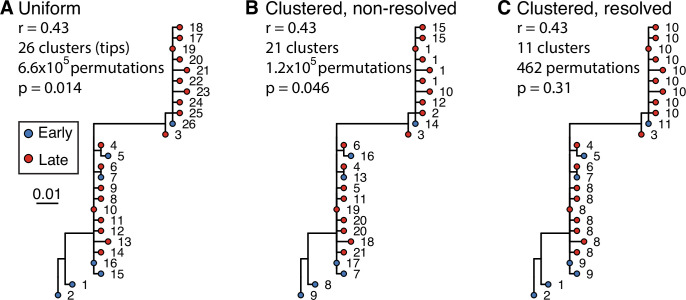
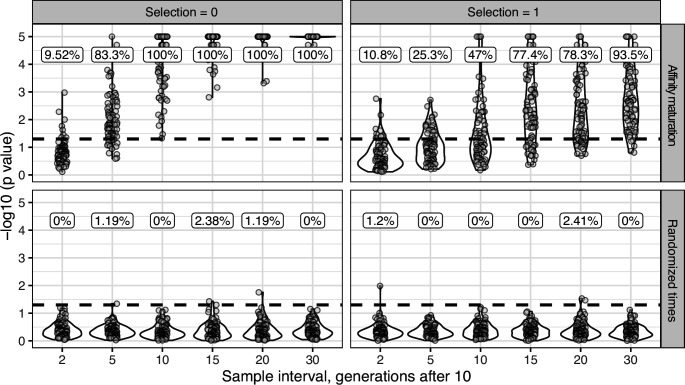
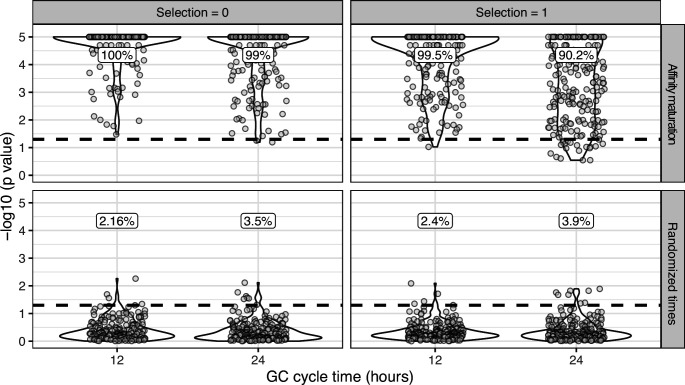
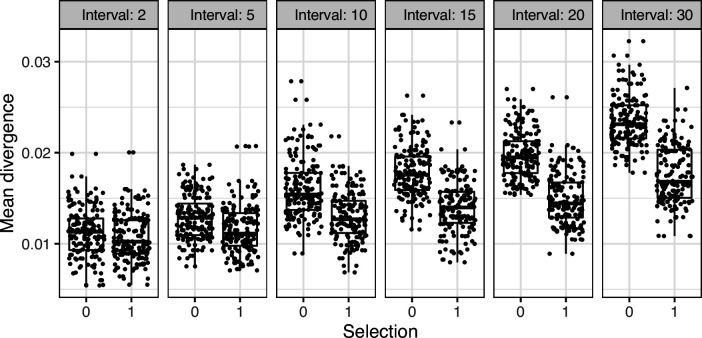
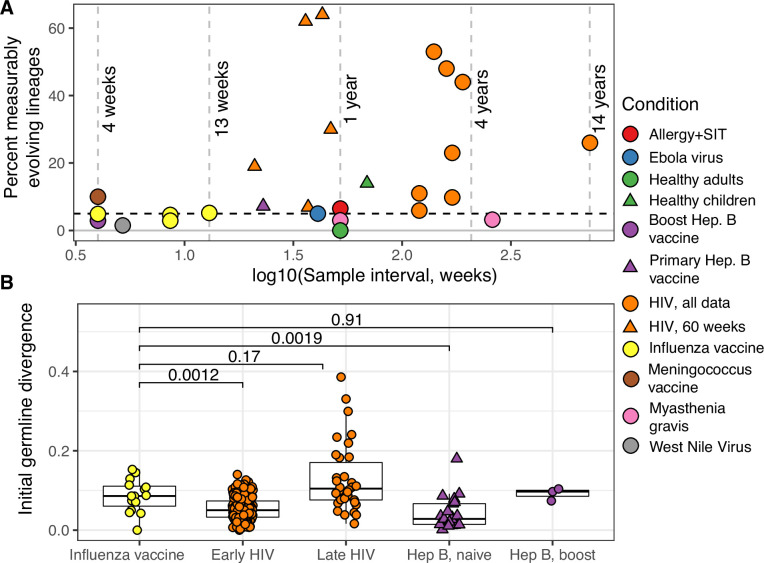
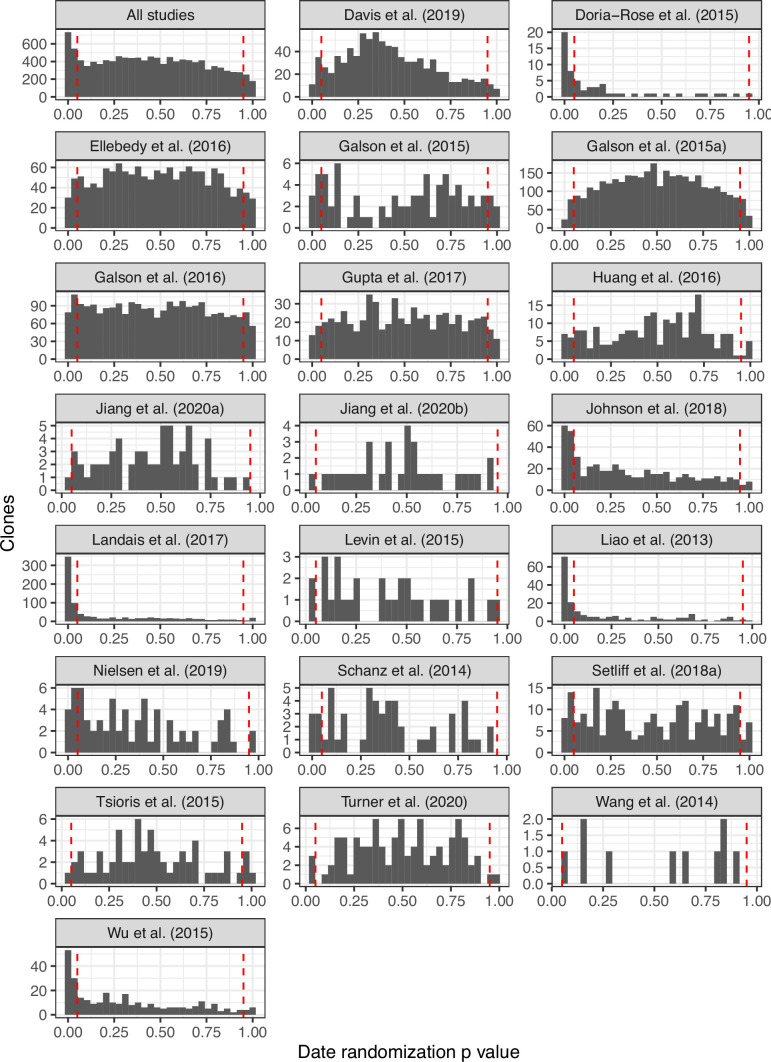
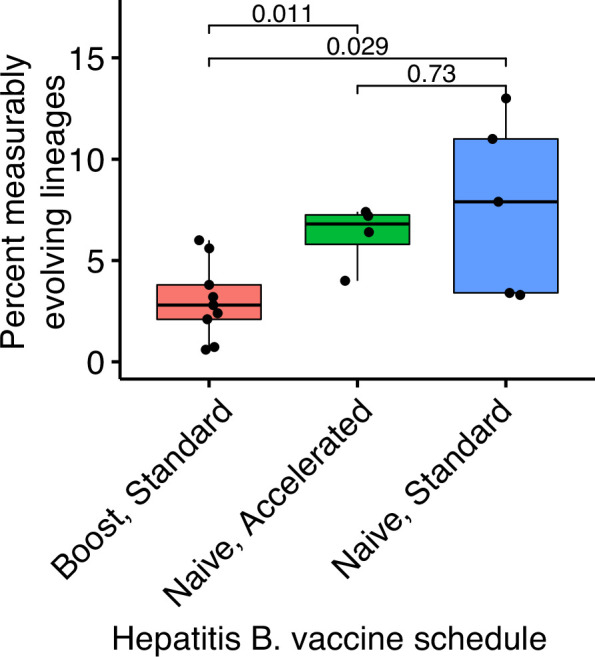
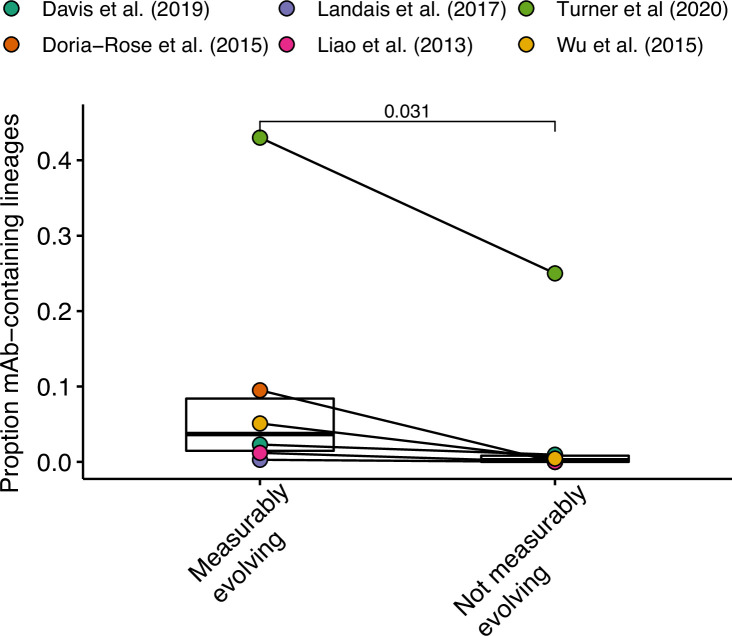
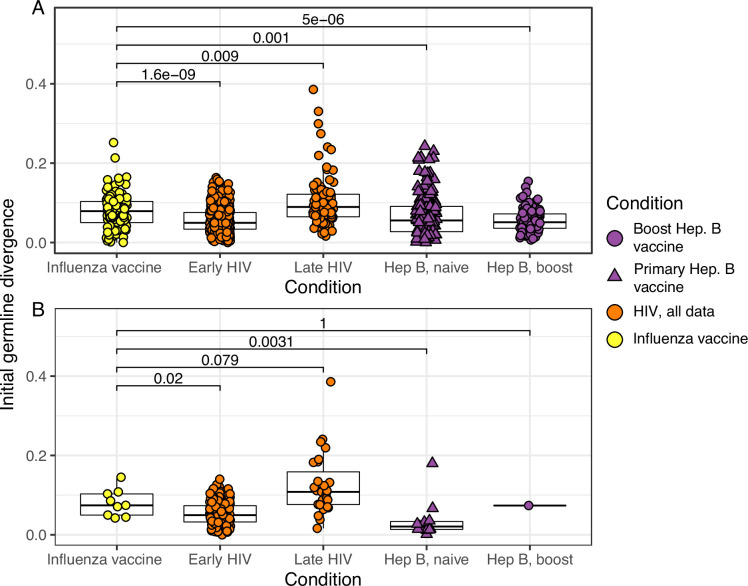
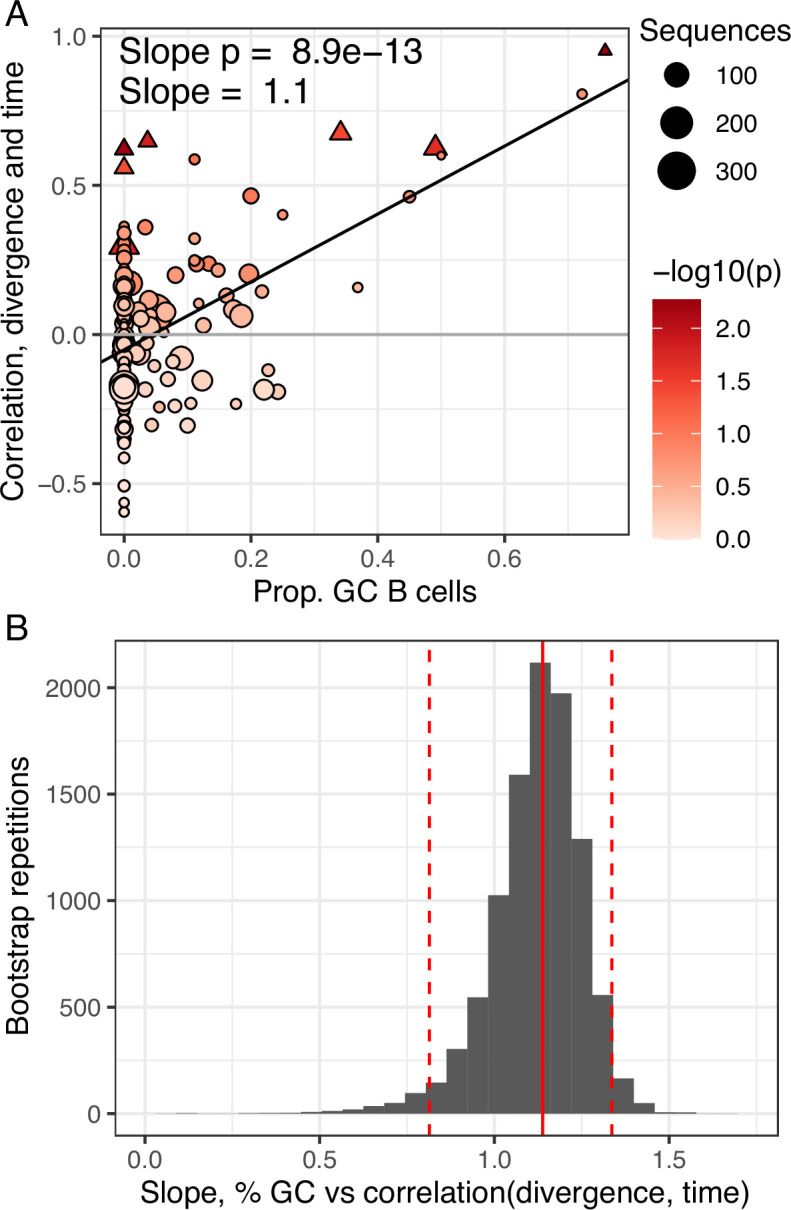
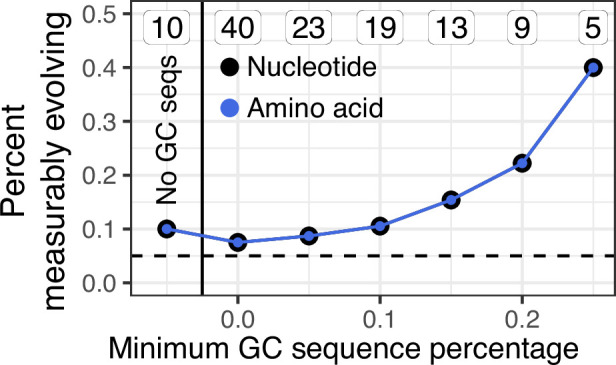
The poor efficacy of seasonal influenza virus vaccines is often attributed to pre-existing immunity interfering with the persistence and maturation of vaccine-induced B cell responses. We previously showed that a subset of vaccine-induced B cell lineages are recruited into germinal centers (GCs) following vaccination, suggesting that affinity maturation of these lineages against vaccine antigens can occur. However, it remains to be determined whether seasonal influenza vaccination stimulates additional evolution of vaccine-specific lineages, and previous work has found no significant increase in somatic hypermutation among influenza-binding lineages sampled from the blood following seasonal vaccination in humans. Here, we investigate this issue using a phylogenetic test of measurable immunoglobulin sequence evolution. We first validate this test through simulations and survey measurable evolution across multiple conditions. We find significant heterogeneity in measurable B cell evolution across conditions, with enrichment in primary response conditions such as HIV infection and early childhood development. We then show that measurable evolution following influenza vaccination is highly compartmentalized: while lineages in the blood are rarely measurably evolving following influenza vaccination, lineages containing GC B cells are frequently measurably evolving. Many of these lineages appear to derive from memory B cells. We conclude from these findings that seasonal influenza virus vaccination can stimulate additional evolution of responding B cell lineages, and imply that the poor efficacy of seasonal influenza vaccination is not due to a complete inhibition of vaccine-specific B cell evolution.
Keywords: B cell; B cell receptor; computational biology; human; immunology; inflammation; measurable evolution; phylogenetics; somatic hypermutation; systems biology; temporal evolution.
Plain language summary
When the immune system encounters a disease-causing pathogen, it releases antibodies that can bind to specific regions of the bacterium or virus and help to clear the infection. These proteins are generated by B cells which, upon detecting the pathogen, can begin to mutate and alter the structure of the antibody they produce: the better the antibody is at binding to the pathogen, the more likely the B cell is to survive. This process of evolution produces B cells that make more effective antibodies. After the infection, some of these cells become ‘memory B cells’ which can be stimulated in to action when the pathogen invades again. Many vaccines also depend on this process to trigger the production of memory B cells that can fight off a specific disease-causing agent. However, it is unclear to what extent memory B cells that already exist are able to continue to evolve and modify their antibodies. This is particularly important for the flu vaccine, as the virus that causes influenza rapidly mutates. To provide high levels of protection, the memory B cells formed following the vaccine may therefore need to evolve to make different antibodies that recognize mutated forms of the virus. It is thought that the low effectiveness of the flu vaccine is partially because the response it triggers does not stimulate additional evolution of memory B cells. To test this theory, Hoehn et al. developed a computational method that can detect the evolution of B cells over time. The tool was applied to samples collected from the blood and lymph nodes (organ where immune cells reside) of people who recently received the flu vaccine. The results were then compared to B cells taken from people after different infections, vaccinations, and other conditions. Hoehn et al. found the degree to which B cells evolve varies significantly between conditions. For example, B cells produced during chronic HIV infections frequently evolved over time, while such evolution was rarely observed during the autoimmune disease myasthenia gravis. The analysis also showed that memory B cells produced by the flu vaccine were able to evolve if recruited to the lymph nodes, but this was rarely detected in B cells in the blood. These findings suggest the low efficacy of the flu vaccine is not due to a complete lack of B cell evolution, but likely due to other factors. For instance, it is possible the evolutionary process it stimulates is not as robust as in other conditions, or is less likely to produce long-lived B cells that release antibodies. More research is needed to explore these ideas and could lead to the development of more effective flu vaccines.
© 2021, Hoehn et al.
Conflict of interest statement
KH receives consulting fees from Prellis Biologics, JT is the recipient of a licensing agreement with Abbvie and has received consulting fees from Gerson Lehman Group, FM, RJ, OP No competing interests declared, AE The Ellebedy laboratory received funding under sponsored research agreements from Emergent BioSolutions and AbbVie, SK receives consulting fees from Northrop Grumman and Peraton
Figures



Similar articles
-
Human germinal centres engage memory and naive B cells after influenza vaccination.Nature. 2020 Oct;586(7827):127-132. doi: 10.1038/s41586-020-2711-0. Epub 2020 Aug 31. Nature. 2020. PMID: 32866963 Free PMC article.
-
Recalling the Future: Immunological Memory Toward Unpredictable Influenza Viruses.Front Immunol. 2019 Jul 2;10:1400. doi: 10.3389/fimmu.2019.01400. eCollection 2019. Front Immunol. 2019. PMID: 31312199 Free PMC article. Review.
-
Flow cytometry reveals that H5N1 vaccination elicits cross-reactive stem-directed antibodies from multiple Ig heavy-chain lineages.J Virol. 2014 Apr;88(8):4047-57. doi: 10.1128/JVI.03422-13. Epub 2014 Feb 5. J Virol. 2014. PMID: 24501410 Free PMC article.
-
Maturation of germinal center B cells after influenza virus vaccination in humans.J Exp Med. 2024 Aug 5;221(8):e20240668. doi: 10.1084/jem.20240668. Epub 2024 Jun 27. J Exp Med. 2024. PMID: 38935072 Free PMC article.
-
Tfh cell response in influenza vaccines in humans: what is visible and what is invisible.Curr Opin Immunol. 2019 Aug;59:9-14. doi: 10.1016/j.coi.2019.02.007. Epub 2019 Mar 25. Curr Opin Immunol. 2019. PMID: 30921542 Free PMC article. Review.
Cited by
-
First Impressions Matter: Immune Imprinting and Antibody Cross-Reactivity in Influenza and SARS-CoV-2.Pathogens. 2023 Jan 21;12(2):169. doi: 10.3390/pathogens12020169. Pathogens. 2023. PMID: 36839441 Free PMC article. Review.
-
Vaccination against rapidly evolving pathogens and the entanglements of memory.Nat Immunol. 2024 Nov;25(11):2015-2023. doi: 10.1038/s41590-024-01970-2. Epub 2024 Oct 9. Nat Immunol. 2024. PMID: 39384979 Review.
-
Inferring B cell phylogenies from paired heavy and light chain BCR sequences with Dowser.bioRxiv [Preprint]. 2023 Oct 2:2023.09.29.560187. doi: 10.1101/2023.09.29.560187. bioRxiv. 2023. Update in: J Immunol. 2024 May 15;212(10):1579-1588. doi: 10.4049/jimmunol.2300851. PMID: 37873135 Free PMC article. Updated. Preprint.
-
B cell phylogenetics in the single cell era.Trends Immunol. 2024 Jan;45(1):62-74. doi: 10.1016/j.it.2023.11.004. Epub 2023 Dec 27. Trends Immunol. 2024. PMID: 38151443 Free PMC article. Review.
-
Vaccination of SARS-CoV-2-infected individuals expands a broad range of clonally diverse affinity-matured B cell lineages.Nat Commun. 2023 Apr 19;14(1):2249. doi: 10.1038/s41467-023-37972-1. Nat Commun. 2023. PMID: 37076511 Free PMC article.
References
-
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
