

Long-Term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-in-human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial
- PMID: 34788571
- PMCID: PMC8817703
- DOI: 10.1089/nat.2021.0043
Long-Term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-in-human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial
Abstract
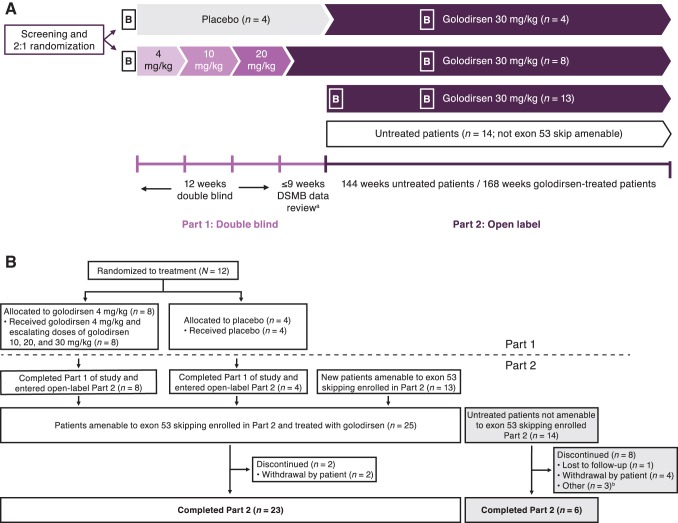
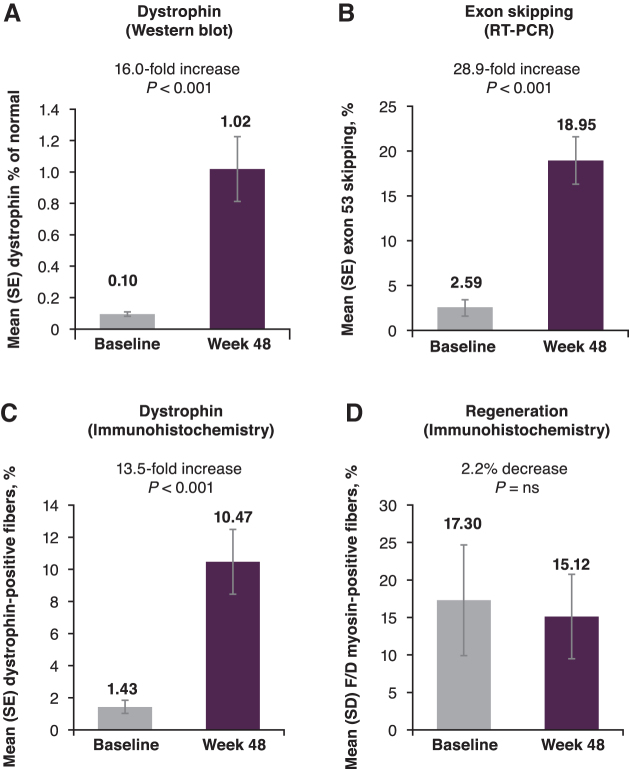
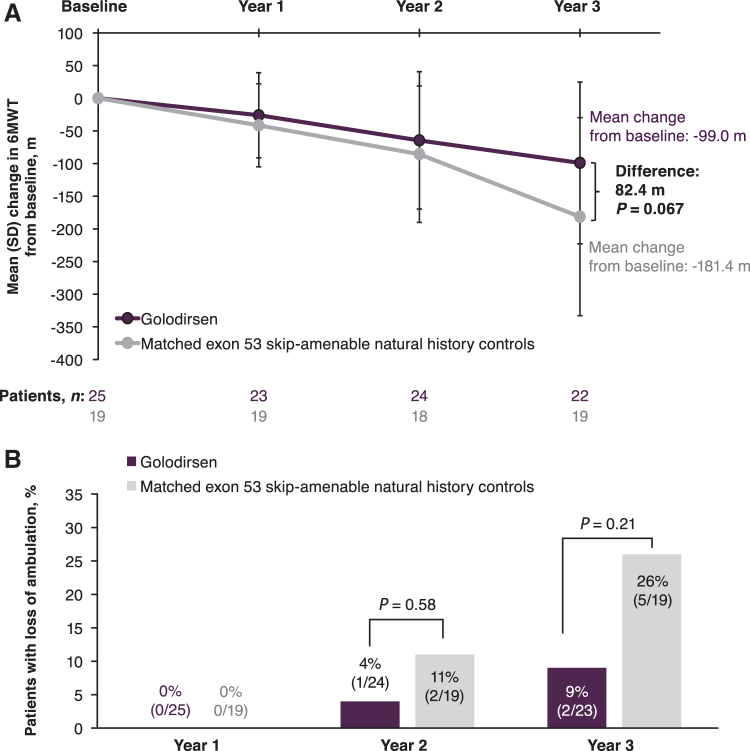
The aim of this Phase 1/2, 2-part, multicenter trial was to report clinical safety and efficacy of long-term golodirsen treatment among ambulatory patients with exon 53 skip-amenable Duchenne muscular dystrophy (DMD). Part 1 was a 12-week, randomized, double-blind, placebo-controlled, dose-titration study followed by 9-week safety review. Part 2 was a 168-week, open-label evaluation of golodirsen 30 mg/kg. Part 1 primary endpoint was safety. Part 2 primary endpoints were dystrophin protein expression and 6-minute walk test (6MWT); secondary endpoints were percent predicted forced vital capacity (FVC%p) and safety. Post hoc ambulation analyses used mutation-matched external natural history controls. All patients from Part 1 (golodirsen, n = 8; placebo, n = 4) plus 13 additional patients entered Part 2; 23 completed the study. Adverse events were generally mild, nonserious, and unrelated to golodirsen, with no safety-related discontinuations or deaths. Golodirsen increased dystrophin protein (16.0-fold; P < 0.001) and exon skipping (28.9-fold; P < 0.001). At 3 years, 6MWT change from baseline was -99.0 m for golodirsen-treated patients versus -181.4 m for external controls (P = 0.067), and loss of ambulation occurred in 9% versus 26% (P = 0.21). FVC%p declined 8.4% over 3 years in golodirsen-treated patients, comparing favorably with literature-reported rates. This study provides evidence for golodirsen biologic activity and long-term safety in a declining DMD population and suggests functional benefit versus external controls. Clinical Trial Registration number: NCT02310906.
Keywords: Duchenne muscular dystrophy; exon skipping; golodirsen.
Conflict of interest statement
L.S. has served on advisory boards for Sarepta Therapeutics, Inc.; E.M. has received consultant fees from Sarepta Therapeutics, Inc.; V.S. has received speaker honoraria from Sanofi Genzyme, is or has recently been on advisory boards for Audentes Therapeutics, Biogen, Exonics Therapeutics/Vertex, Novartis, Roche, Sarepta Therapeutics, Inc., and Wave Therapeutics, and has research collaborations with Sanofi Genzyme and Ultragenyx; M.G. has received speaker honoraria from Sarepta Therapeutics, Inc., is on advisory boards for Pfizer, has research collaboration with Sarepta Therapeutics, Inc., and is the Chair of the VBP15-004 study but does not have any financial interest with ReveraGen; A.M.S., M.S., and D.L. have nothing to disclose; E.K., N.K., A.D., X.W., B.H., and D.W. are or have been employees of Sarepta Therapeutics, Inc.; F.M. has received consultant fees and speaker honoraria from Sarepta Therapeutics, Inc., and is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre.
Figures
References
-
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, et al. (2018). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 17:251–267. - PMC - PubMed
-
- Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R and Bushby K. (2002). Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord 12:926–929. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical