

Mutations of the histone linker H1-4 in neurodevelopmental disorders and functional characterization of neurons expressing C-terminus frameshift mutant H1.4
- PMID: 34788807
- PMCID: PMC9271223
- DOI: 10.1093/hmg/ddab321
Mutations of the histone linker H1-4 in neurodevelopmental disorders and functional characterization of neurons expressing C-terminus frameshift mutant H1.4
Abstract
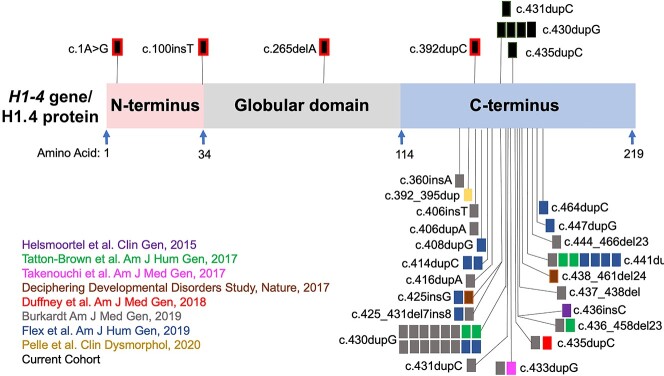
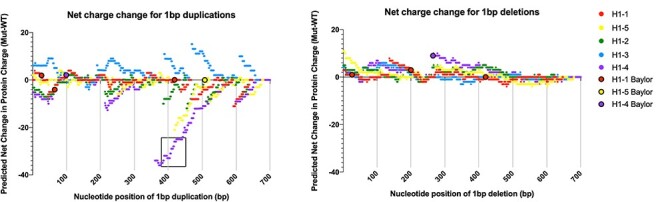
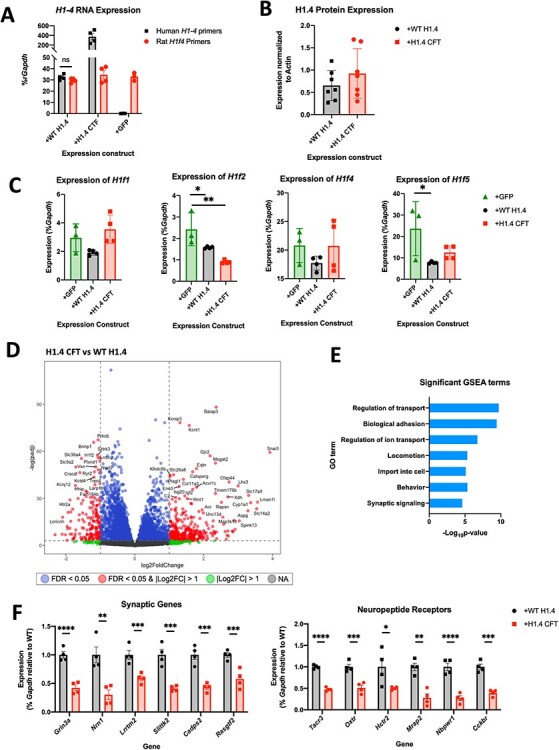
Rahman syndrome (RMNS) is a rare genetic disorder characterized by mild to severe intellectual disability, hypotonia, anxiety, autism spectrum disorder, vision problems, bone abnormalities and dysmorphic facies. RMNS is caused by de novo heterozygous mutations in the histone linker gene H1-4; however, mechanisms underlying impaired neurodevelopment in RMNS are not understood. All reported mutations associated with RMNS in H1-4 are small insertions or deletions that create a shared frameshift, resulting in a H1.4 protein that is both truncated and possessing an abnormal C-terminus frameshifted tail (H1.4 CFT). To expand understanding of mutations and phenotypes associated with mutant H1-4, we identified new variants at both the C- and N-terminus of H1.4. The clinical features of mutations identified at the C-terminus are consistent with other reports and strengthen the support of pathogenicity of H1.4 CFT. To understand how H1.4 CFT may disrupt brain function, we exogenously expressed wild-type or H1.4 CFT protein in rat hippocampal neurons and assessed neuronal structure and function. Genome-wide transcriptome analysis revealed ~ 400 genes altered in the presence of H1.4 CFT. Neuronal genes downregulated by H1.4 CFT were enriched for functional categories involved in synaptic communication and neuropeptide signaling. Neurons expressing H1.4 CFT also showed reduced neuronal activity on multielectrode arrays. These data are the first to characterize the transcriptional and functional consequence of H1.4 CFT in neurons. Our data provide insight into causes of neurodevelopmental impairments associated with frameshift mutations in the C-terminus of H1.4 and highlight the need for future studies on the function of histone H1.4 in neurons.
© The Author(s) 2021. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures



References
-
- Burkardt, D.D., Zachariou, A., Loveday, C., Allen, C.L., Amor, D.J., Ardissone, A., Banka, S., Bourgois, A., Coubes, C., Cytrynbaum, C.et al. (2019) HIST1H1E heterozygous protein-truncating variants cause a recognizable syndrome with intellectual disability and distinctive facial gestalt: a study to clarify the HIST1H1E syndrome phenotype in 30 individuals. Am. J. Med. Genet. A, 179, 2049–2055. - PubMed
-
- Duffney, L.J., Valdez, P., Tremblay, M.W., Cao, X., Montgomery, S., McConkie-Rosell, A. and Jiang, Y.H. (2018) Epigenetics and autism spectrum disorder: a report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am. J. Med. Genet. B Neuropsychiatr. Genet., 177, 426–433. - PMC - PubMed
-
- Takenouchi, T., Uehara, T., Kosaki, K. and Mizuno, S. (2018) Growth pattern of Rahman syndrome. Am. J. Med. Genet. A, 176, 712–714. - PubMed
-
- Flex, E., Martinelli, S., Van Dijck, A., Ciolfi, A., Cecchetti, S., Coluzzi, E., Pannone, L., Andreoli, C., Radio, F.C., Pizzi, S.et al. (2019) Aberrant function of the C-terminal tail of HIST1H1E accelerates cellular senescence and causes premature aging. Am. J. Hum. Genet., 105, 493–508. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
